
Blum, K., Baron, D., McLaughlin, T., & Gold, M. S. (2020). Molecular neurological correlates of endorphinergic/dopaminergic mechanisms in reward circuitry linked to endorphinergic deficiency syndrome (EDS). Journal of the neurological sciences, 411, 116733.
Points forts
- La dopamine mésolimbique est liée à la motivation, à la lutte contre le stress, à la perception des incitations et au bien-être.
- Déficit de récompense ; déficit en neurotransmetteurs dû à une déficience génétique/épigénétique.
- Consommation chronique de substances psychoactives – Régulation négative – Anhédonie et comportements de recherche.
- Trouble de l’usage des opioïdes, maintenant traitement de substitution aux opioïdes à vie pour la réduction des risques.
- Proposition d’induction de l’homéostasie dopaminergique nécessaire et du bien-être à vie.
- Identifier les déficits en neurotransmetteurs et les restaurer avec des nutraceutiques de précision.
Abstract
Le consensus de la littérature actuelle soutient fortement le concept selon lequel les neurotransmetteurs cérébraux et les seconds messagers impliqués dans la libération nette de dopamine dans la région mésolimbique, en particulier le Nucleus Accumbens (NAc), sont directement liés à la motivation, à l’anti-stress, à la saillance de l’incitation (désir) et au bien-être. Le rôle de la dopamine dans la symptomatologie du sevrage alcoolique, le comportement de manque de cocaïne, les produits de condensation de la dopamine (TIQ) et, plus récemment, les aspects génétiques de la recherche de drogues et la régulation de la pro-dopamine, fournissent des preuves irréfutables des corrélats neurologiques moléculaires pertinents des mécanismes dopaminergiques/endorphinergiques dans le circuit de la récompense en raison de polymorphismes génétiques et d’insultes épigénétiques. Face à l’épidémie d’opioïdes des Américains, le consensus clinique consiste à traiter le trouble de l’utilisation des opioïdes (OUD) avec un traitement de substitution aux opioïdes à vie. Cependant, les auteurs suggèrent un changement de paradigme impliquant de nouvelles modalités telles que le ciblage du système endorphinergique lié à la libération de dopamine dans le NAc, en termes d’induction de l'”homéostasie dopaminergique” requise. En s’appuyant sur le théorème connu de l’interaction génétique-environnement P = G +E, les auteurs justifient clairement l’adoption d’un test de risque génétique associé à une régulation endorphinergique/dopaminergique pour remédier au dysfonctionnement de l’ensemble du circuit cérébral de la récompense. L’objectif de modifier la connectivité fonctionnelle à l’état de repos peut nécessiter une “correction des neurotransmetteurs” par l’inhibition de l’enképhalinase pour surmonter ou combattre l’auto-induction de la libération aiguë de dopamine par l’abus de substances psychoactives, qui entraîne une régulation chronique de la dopamine à la baisse. En tant que sous-ensembles du déficit de récompense, nous sommes prêts à fournir une nouvelle thérapie génétiquement guidée pour les déficiences endorphinergiques, opioïdes et dopaminergiques et les syndromes associés, en utilisant la “gestion de précision de la dépendance”.
1. Introduction
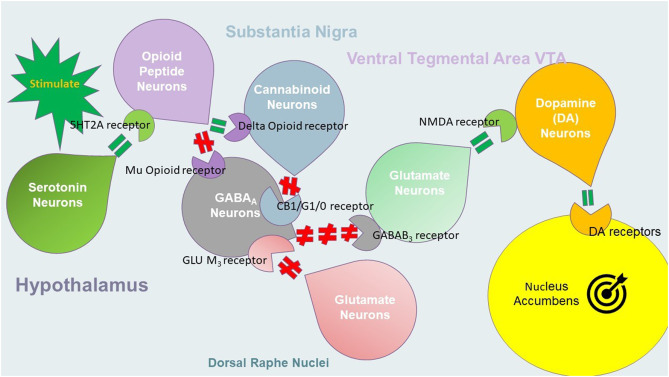
Plusieurs neurotransmetteurs sont impliqués dans le traitement de la récompense et de la punition. Ces voies impliquent au moins six neurotransmetteurs majeurs et de nombreux seconds messagers, liés au mésolimbique et au cortex préfrontal (PFC). L’une des fonctions est de réguler la voie finale du “désir”, en provoquant une libération nette de dopamine dans les neurones [1]. La figure 1 présente une représentation schématique de la cascade de récompense cérébrale (CRC) montrant l’interaction des systèmes sérotoninergique, cannabinoidergique, opioidergique, GABAergique, glutaminergique et dopaminergique liés à la libération nette de dopamine dans le Nucleus Accumbens (NAc). Dans cet article, les auteurs mettent l’accent sur la dopamine, sachant que le traitement sain d’un potentiel d’action initial dans le cerveau nécessite l’intégrité de l’ensemble du complexe de neurotransmetteurs du circuit cérébral de la récompense. Les interactions en cascade aboutissent à une libération équilibrée de dopamine, non seulement dans le NAc, mais aussi dans de nombreuses régions du cerveau. Ces régions sont impliquées dans la motivation, la cognition (mémoire), le plaisir, la réduction du stress, la prise de décision, le rappel, la réintégration des drogues, les envies et le bien-être. Le résultat est de fournir à l’Homo sapiens un point de consigne de bonheur sain ainsi qu’une connectivité fonctionnelle à l’état de repos [2].
La fonction dopaminergique neurologique dans une perspective historique
La dopamine (DA 3,4-dihydroxyphénéthylamine), nommée d’après le précurseur de la lévodopa, la L-DOPA, est formée dans le cerveau et les reins. La dopamine est une catécholamine de la famille des phénéthylamines. Le cerveau possède plusieurs voies dopaminergiques distinctes, notamment celles qui impliquent le contrôle de la motricité, la libération de diverses hormones et la neurotransmission.
La culture populaire et les médias décrivent souvent la dopamine comme la principale substance chimique du plaisir ; cependant, sur la base de recherches sur les animaux, l’opinion actuelle est que la dopamine confère plutôt une saillance motivationnelle [1,2]. La dopamine joue un rôle essentiel dans la motivation comportementale. La libération de dopamine indique l’importance de la motivation, la désirabilité ou l’aversion d’un résultat, et affecte le comportement vers ou loin de ce résultat [3]. Le niveau de dopamine dans le cerveau augmente avec l’anticipation de la plupart des types de récompenses, et de nombreuses drogues addictives connues, comme les opioïdes, augmentent de manière aiguë la libération de dopamine ou bloquent sa recapture dans les neurones après la libération. Cependant, en cas de dépendance chronique, c’est l’inverse qui se produit et, en fait, dans ces cas, la fonction dopaminergique est régulée à la baisse [[4], [5], [6], [7]]]. Des études pertinentes et instructives réalisées par Uhl, Koob et Cable [6] et Elman et al. [5] appuient ces concepts.
La dopamine a été synthétisée pour la première fois par George Berger et James Ewens aux laboratoires Wellcome de Londres, en Angleterre [8], en 1910, bien avant que Kathleen Montagu ne la localise dans le cerveau humain en 1957. La fonction de neurotransmetteur de la dopamine a été reconnue un an plus tard par Arvid Carlsson et Nils-Ake Hillarp. Carlsson, Paul Greengard et Eric Kandel ont reçu le prix Nobel 2000 de physiologie ou de médecine pour leurs découvertes concernant la transduction du signal de la dopamine dans le système nerveux et son implication dans l’apprentissage et la mémoire. Chez l’homme, la dopamine se lie aux récepteurs de la surface cellulaire et les active. La dopamine a une forte affinité de liaison avec les sous-types D1 à D5 des récepteurs de la dopamine et avec le récepteur 1 associé aux traces d’amines chez l’homme (hTAAR1) [9,10]. La dopamine fonctionne également comme une protéine métabotropique couplée à la protéine G (protéine liant les nucléotides de guanine). Lorsque la dopamine est liée aux récepteurs, ceux-ci exercent des réponses cellulaires par l’intermédiaire d’un système complexe de second messager [11], qui détecte les molécules extérieures à la cellule et active les voies de transduction du signal interne.
La dopamine et la récompense (un aperçu)
Les événements gratifiants activent les neurones dopaminergiques de la substantia nigra et de la VTA, comme le montrent les enregistrements de signaux neuronaux effectués dans le cerveau d’animaux [12]. Ces neurones dopaminergiques sensibles à la récompense sont essentiels à la cognition liée à la récompense et constituent l’élément central du système de récompense. La fonction de la dopamine est différente dans chaque projection axonale. Par exemple, les projections de la coquille de la VTA et de la substantia nigra attribuent une saillance incitative (“vouloir”) aux indices de stimuli gratifiants, tandis que les projections du cortex orbitofrontal de la VTA évaluent différents objectifs comportementaux en fonction de l’importance de l’incitation. Les projections de l’amygdale et de la VTA hippocampique interviennent dans la consolidation des souvenirs liés à la récompense, et les voies VTA-NAc core et substantia nigra- striatum dorsal sont impliquées dans l’apprentissage de réponses motrices qui facilitent l’acquisition de stimuli gratifiants. En outre, une certaine activité au sein des projections dopaminergiques de la VTA semble être associée à la prédiction de la récompense [[13], [14], [15]]].
Si la dopamine joue un rôle central dans le “désir”, elle est également associée aux réponses comportementales appétitives ou d’approche des stimuli gratifiants, qui se traduisent par une réponse comportementale de consommation, et de nombreuses expériences animales révèlent que la dopamine n’est pas synonyme de “goût” ou de plaisir hédoniste. L’implication de la dopamine dans le plaisir et la récompense fait l’objet d’une controverse qui a entraîné une certaine confusion sur la manière de distinguer la motivation du plaisir, ainsi que le “vouloir” du “plaire”. Sousa et al [16] ont démontré que le système cérébral semble avoir évolué différemment chez l’homme et chez les singes. Ils ont indiqué que les études sur les animaux ne sont pas les mêmes que les informations cliniques décrites par les auto-évaluations chez les humains. Leurs conclusions suggèrent également que les données ne permettent pas de se fier entièrement aux extrapolations des études sur les rongeurs et les primates non humains. En ce qui concerne les concepts de plaisir, de dopamine et de renforcement, l’expérience humaine est plus importante que les données animales.
Néanmoins, les données animales ont de la valeur et l’utilisation de modèles animaux doit être encouragée, tout en mettant en garde contre l’interprétation des résultats, sans sauter à des conclusions qui peuvent être expliquées par des données provenant d’expériences humaines de suivi [16,17]. Une étude clinique réalisée en janvier 2019 a évalué l’effet du précurseur de la dopamine (L-DOPA), de l’antagoniste (rispéridone) et du placebo sur les réponses de récompense à la musique. Ferreri et al [18] ont mesuré les changements dans l’activité électrodermale pour évaluer le degré de plaisir ressenti pendant les frissons musicaux, ainsi que les évaluations subjectives. Ils ont constaté que la manipulation de la neurotransmission de la dopamine régule la cognition du plaisir (en particulier, l’impact hédonique de la musique) de manière bidirectionnelle chez les sujets humains. Les auteurs suggèrent qu’une neurotransmission dopaminergique accrue est une condition sine qua non pour des réactions hédoniques agréables à la musique chez l’homme [18].
Dopamine et comportement addictif
L’addiction est un système cérébral de récompense perturbé, acquis par des mécanismes épigénétiques et une expression transcriptionnelle chronique de l’ARNm. L’exposition chronique à des stimuli addictifs entraîne un engagement incontrôlable avec des stimuli gratifiants, quelles que soient les conséquences négatives. Les stimuli naturels tels que la nourriture et l’exercice, ainsi que les comportements à forte dose d’excitation, tels que les jeux d’argent et de hasard, et les substances, telles que les opioïdes, induisent tous la neuroplasticité [5,[19],[20],[21]].
La reconnaissance du rôle des polymorphismes de l’ADN, notamment en ce qui concerne la fonction dopaminergique, est tout aussi importante [22,23]. Le traitement de la symptomatologie chronique tertiaire induite par les drogues est au mieux à courte vue si on le compare à la prise en compte de nouvelles cibles épidémiologiques pour la prophylaxie à long terme liée à des interventions précoces. Par exemple, Delta FosB (ΔFosB), un facteur de transcription génétique, est un médiateur central dans le développement épigénétique de toutes les dépendances aux drogues et autres [[24], [25], [26], [27]]]. Sur la base d’une revue intensive de la littérature, ΔFosB module la compulsivité au cours de l’acquisition de l’addiction [[28,29]. La surexpression de ΔFosB dans les neurones à épines moyennes de type D1 du noyau accumbens régule l’auto-administration de drogues et la sensibilisation à la récompense, directement, par le biais du renforcement positif tout en atténuant la sensibilité à l’aversion [30].
Identification des neurotransmetteurs et de leur fonction
En 1921, un scientifique autrichien nommé Otto Loewi a découvert le premier neurotransmetteur important, l’acétylcholine (AN), suivi d’autres neurotransmetteurs primaires, tels que la sérotonine, la glutamine, les enképhalines et la dopamine. Leurs fonctions neurologiques ont été découvertes au cours du 20e siècle, les récepteurs de type opioïde n’ayant été identifiés qu’au milieu des années 70 [31], tandis que le rôle potentiel de la sérotonine dans la consommation d’alcool, découvert par le groupe Myers à la fin des années 60, a été l’une des principales découvertes du nouveau domaine des neurosciences [32]. Blum a rapporté les effets de la dopamine et d’autres catécholamines sur la transmission neuromusculaire en 1969. Cette découverte a fourni les premières preuves du rôle de la dopamine et des tremblements dans la maladie de Parkinson [33].
Corrélats endorphinergiques du traitement de la récompense
Les endorphines ont été découvertes en 1974 par deux groupes de chercheurs indépendants. Hughes et Kosterlitz ont isolé les enképhalines dans le cerveau de porc [34], et Eric Simon a découvert des récepteurs opioïdes dans le cerveau des vertébrés [35]. Les endorphines sont des neuropeptides opioïdes endogènes produits par le système nerveux central (CNS) et l’hypophyse des animaux et de l’Homo sapiens. Il est important de noter que ces peptides sont classés en trois catégories – enképhalines, endorphines et dynorphines – dérivées de trois molécules précurseurs distinctes (Pro- enképhaline A, Pro-Endorphine et Pro-dynorphine ou Pro- enképhaline B). Les peptides opioïdes fonctionnels et actifs sont générés à partir de précurseurs nés dans le cerveau par des enzymes appelées peptidases. Ces peptidases clivent (coupent) des molécules précurseurs spécifiques en molécules plus petites. Des endorphines telles que la bêta-endorphine (contenant 91 acides aminés) et des molécules plus petites telles que la leu-enképhaline et la met-enképhaline, qui ne contiennent que cinq acides aminés (pentapeptides). S’il existe des endorphines de type alpha, delta, sigma et gamma aux fonctions sélectives dans le cerveau et les tissus périphériques, dérivées de peptides connus plus importants, comme la POMC [285 acides aminés], il existe également d’autres ligands connus sous le nom de dynorphines : la dynorphine A, la dynorphine B et la α/β néoendorphine, dérivées de la molécule plus importante synthétisée dans le cerveau. Les dynorphines sont les principaux ligands du récepteur opioïde Kappa, mais elles ont également une faible affinité pour d’autres récepteurs opioïdes, notamment delta et mu [36]. La classe des endorphines comprend trois composés – les alpha-endorphines, les bêta-endorphines et les gamma-endorphines – qui se lient de préférence aux récepteurs opioïdes [37]. L’inhibition de la signalisation de la douleur est la principale fonction des endorphines. Elles produisent également une sensation d’euphorie très similaire à celle produite par d’autres opioïdes via la libération de dopamine [38]. Diverses activités humaines déclenchent la production d’endorphines. Par exemple, le rire, connu pour stimuler la production d’endorphines, augmente la résistance à la douleur [39], et la β-endorphine est libérée lors d’un exercice aérobique vigoureux [40,41]. En effet, de nombreuses activités agréables, y compris la consommation de nourriture et de chocolat, qui contiennent de la tétrahydroisoquinoline (TIQ), ainsi que le sexe, l’orgasme, le yoga, la méditation et l’écoute de la musique, entraînent la libération d’endorphines [41]. Des études ont également démontré que les tissus humains et divers tissus animaux sont capables de produire de la morphine, qui n’est pas un peptide [42].
Les enképhalines sont très présentes dans le cerveau et ce polypeptide est lié au fonctionnement du cerveau en cas de réponse au stress, en particulier dans l’hippocampe et le cortex préfrontal. Les facteurs de stress ont un impact sur les neuropeptides et leur action est métabotropique dans des régions spécifiques du cerveau [43]. Enfin, les récepteurs des enképhalines sont couplés aux protéines G, y compris les récepteurs delta-opioïdes et mu. D’autres ligands opioïdes endogènes, tels que les dynorphines, se lient aux récepteurs kappa, et les endorphines elles-mêmes se lient aux récepteurs mu [44].
Dans le système de récompense mésolimbique, les enképhalines inhibent la libération du neurotransmetteur GABA lorsqu’elles se lient au récepteur μ et augmentent la production et la libération de dopamine [45].
Cette brève revue de l’identification des neurotransmetteurs et de leur fonction dans le circuit cérébral de la récompense, souligne que le risque génétique pour tous les comportements addictifs avec ou sans substance, posé par une neurotransmission défectueuse attribuable à des polymorphismes génétiques et appelé syndrome de déficience de récompense (RDS) [10,23,46]. Le tableau 1 énumère les comportements addictifs qui ont été identifiés comme des comportements RDS.
Dévoiler les fondements neurologiques et mettre l’accent sur le traitement des causes profondes de la dépendance au lieu de la prolonger toute la vie.
Les termes “Recovery” et “The 12-Step Program and Fellowship” sont devenus des noms familiers au début des années 1980 [47]. L’American Society of Addiction Medicine (ASAM) a introduit une nouvelle définition de la “dépendance” en tant que trouble cérébral en 2011 [48], l’acceptation de cette idée est maintenant bien établie et a eu un impact positif sur la recherche fondamentale [49]. Le travail de Blum avec Ernest P. Noble, la découverte du premier gène associé à l’alcoolisme sévère, a conduit au domaine actuel de la “génétique psychiatrique” [50]. Aujourd’hui, au XXIe siècle, la science des addictions, grâce à l’ère de la médecine génomique, est sur le point de commencer à comprendre la véritable nature de ce trouble cérébral, le “syndrome de déficience de la récompense” [10]. Grâce à ces nombreuses années de recherche et de réflexion, plusieurs exemples de progrès ont été brièvement présentés ici. Il s’agit notamment de la compréhension de :
- les mécanismes neurochimiques impliqués dans le processus de dépendance, y compris la symptomatologie du sevrage,
- les bases physiologiques de la neurotransmission cérébrale et les mécanismes neurochimiques de la fonction synaptique,
- les mécanismes de libération, de catabolisme et de stockage des neurotransmetteurs dans les lieux pré et postsynaptiques,
- le rôle de la potentialisation à long terme dans l’auto-administration de drogues et la sensibilisation,
- la “cascade de récompense cérébrale” et son rôle dans le point de consigne du bonheur et dans le comportement de manque et de rechute,
- et le rôle de la neurogénétique dans tous les aspects de la recherche de drogues et des processus de dépendance.
La recherche actuelle a-t-elle rattrapé ces progrès ? En ce qui concerne cette question, il semble raisonnable, sur la base de mécanismes physiologiques bien connus, que la communauté des médecins spécialistes des addictions ne jette pas le bébé avec l’eau du bain [51]. Le trouble cérébral connu sous le nom de RDS est très compliqué. Les difficultés rencontrées par plusieurs études d’association à l’échelle du génome (GWAS) pour trouver des associations importantes et significatives avec divers gènes peuvent être dues au fait que le trouble est polygénique et, surtout, à l’utilisation erronée de contrôles apparemment inappropriés. Il convient d’examiner la proposition selon laquelle le phénotype actuel de la “dépendance” comprend plusieurs sous-types de SDR [voir tableau 1], y compris, par exemple, les syndromes sérotoninergiques, cannabinoidergiques, endorphinergiques, opioidergiques, glutaminergiques et les syndromes de déficience dopaminergique.
En supposant que la proposition de phénotype du RDS soit correcte, un dépistage rigoureux des sous-types de RDS chez les témoins devrait être effectué avant toute analyse génétique, candidate ou GWAS. La crainte est que la présence de la maladie parmi les témoins n’aboutisse à des résultats erronés et inutiles [52]. La dissection de cette question expérimentale pourrait prendre des années.
Le test GARS (Genetic Addiction Risk Score)
Se pencher sur la gestion clinique de ces allèles de risque polymorphes, qu’il s’agisse de l’induction neurochimique hyperdopaminergique ou hypodopaminergique de l’homéostasie des neurotransmetteurs, semble être un objectif de traitement raisonnable. Les patients (surtout lorsqu’ils sont jeunes) obligés de se rendre dans un centre de traitement (en raison de l’intervention du tribunal, de la famille ou d’amis) peuvent être dans le déni de leurs problèmes comportementaux actuels. Le Genetic Addiction Risk Score (GARS) non invasif [53] est un panel génétique polymorphe qui permet de stratifier le risque génétique chez un individu. Il s’agit essentiellement d’un “miroir de la fonction de récompense du cerveau” qui peut réduire les devinettes et aider à résoudre le déni, la culpabilité et la honte, en démontrant une prédisposition génétique à la consommation de drogues. Le test GARS présente également d’autres avantages cliniques. Ces avantages comprennent le suivi médical des réponses pharmacogénétiques d’un médicament, les questions métaboliques liées à l’administration de la drogue, un traitement personnalisé sur mesure (régulation de la pro-dopamine avec le nutraceutique KB220) ; et la nécessité médicale pour le type de soins cliniques, qui implique l’identification du risque de comportements RDS, et le traitement pharmacogénomique ciblant les polymorphismes des gènes. Un autre avantage substantiel est que la curiosité de la famille concernant le couplage du GARS et de la régulation de la pro-dopamine, a renforcé la volonté de la famille de participer au plan de rétablissement du patient [54]. L’ajout du test génétique aux analyses d’urine afin d’évaluer l’observance des traitements médicamenteux approuvés par la FDA, tels que la Suboxone et la méthadone, et l’utilisation de la Comprehensive Analysis of Reported Drugs (CARD) pour documenter l’abstinence de drogues illicites, psychoactives et addictives, pourraient constituer un élément précieux du plan de traitement des troubles liés à l’utilisation de substances (TUS).
Plus précisément, l’élaboration d’une approche thérapeutique organisée impliquant le matériel susmentionné, concernant, par exemple, le syndrome de carence en opioïdes (SDA), semble la plus productive. Tout d’abord, la compréhension du BRC (Fig. 1) facilite la proposition selon laquelle la libération nette de dopamine au niveau du NAc nécessite une fonction interactive des neurotransmetteurs au niveau du système de récompense mésolimbique du cerveau. Toute interférence dans ce processus se traduira par un manque d’interaction entre les neurotransmetteurs depuis l’hypothalamus jusqu’aux noyaux du raphé, à la substania nigra et, ensuite, à la VTA et au NAc. Une perturbation au sein du CRB peut ensuite entraîner une atténuation de la libération de dopamine, provoquant une carence en dopamine et résultant d’une atteinte génétique [53] ou épigénétique [55], reliant la méthylation des récepteurs dopaminergiques D2 à une vie de comportements addictifs. Des études pertinentes et instructives réalisées par Uhl, Koob et Cable [6] et Elman et al. [5] soutiennent ces concepts.
Cette proposition peut être illustrée en mettant brièvement en évidence les diverses perturbations du CRB causées par les polymorphismes associés à certains allèles à risque mesurés dans le test GARS. Ces polymorphismes génétiques favorisent donc le RDS. Une recherche bibliographique portant sur des études cas-témoins révèle une association significative entre les fonctions de ces gènes et les allèles à risque (voir tableau 2).
Les auteurs affirment qu’une prise en compte appropriée des déficiences en neurotransmetteurs (opioïdes/dopaminergiques) dans le domaine, si elle est adoptée, permettrait d’éliminer de nombreuses séquelles cliniques indésirables et de réduire la confusion concernant le rôle de la génétique dans la toxicomanie.
Sur la base des résultats de la littérature antérieure, bien que non représentatif de toutes les études d’association connues à ce jour, cet échantillon d’études cas-témoins montre des associations significatives entre le risque d’alcoolisme et de toxicomanie. Plusieurs méta-analyses portant sur 110 241 cas et 122 525 témoins révèlent des associations significatives entre les allèles de risque d’alcoolisme et de toxicomanie.
Pour comprendre les mécanismes neurologiques impliqués dans les déficits enképhalinergiques et opioïdes, le rôle des opioïdes endogènes et des récepteurs de type opioïde mérite l’attention. Par exemple, Margolis et al [56] soulignent que la VTA joue un rôle central à la fois dans la motivation et le renforcement. Des agonistes connus des récepteurs kappa et mu-opioïdes (KOP-R et MOP-R) micro-injectés dans la VTA induisent des actions motivationnelles puissantes et opposées. La transmission du glutamate dans la VTA contribue à ces effets motivationnels. Les agonistes des récepteurs opioïdes mu (MOR) ne produisent pas seulement un soulagement de la douleur, mais ont des effets de récompense qui sont liés aux voies dopaminergiques mésolimbiques [56,57].
En outre, Margolis et ses collaborateurs [58] ont constaté que l’activité du récepteur delta-opioïde (DOR) dans la VTA atténuait la consommation d’alcool chez les rats. Cet effet, comme prévu, a été bloqué par la bicuculline, un antagoniste du GABA(A), peut-être parce que la réduction du contrôle inhibiteur du GABA permet la libération accrue de dopamine dans le NAc. De même, Mitchell et al [59] ont montré que, comme prévu, la consommation aiguë d’alcool, même chez les gros buveurs, induit la libération d’opioïdes dans le NAc et le cortex orbitofrontal (OFC), deux zones du cerveau liées à la récompense. En revanche, la consommation chronique d’alcool devrait avoir des effets opposés, notamment la régulation à la baisse de la synthèse des endorphines et la carence en endorphines qui s’ensuit [60]. Hjelmstad et ses collaborateurs [61] ont montré que les opioïdes entraînent une atténuation de l’entrée GABAergique dans les neurones de la VTA, ce qui induit une libération neuronale de dopamine dans le NAc. Par conséquent, les opioïdes endogènes ou exogènes peuvent activer les neurones de la VTA, y compris les neurones DA, comme cela a été observé avec l’optogénétique. En utilisant le radioligand sélectif des récepteurs mu-opioïdes [11C] Carfentanil, Mitchell et al [62] ont également constaté qu’après une consommation aiguë d’alcool, les sujets porteurs de l’allèle COMT Val158 libèrent davantage d’opioïdes dans le NAc droit mais moins dans l’OFC médian, ce qui suggère une régulation génétique de la dopamine via la libération endorphinergique au niveau du site de récompense. En ce qui concerne le soulagement de la douleur, Navratilova et al [63] ont constaté que l’activation pharmacologique des récepteurs opioïdes du gyrus cingulaire antérieur (ACG) droit d’animaux blessés, mais non indolores, stimulait la libération de dopamine dans le NAc et produisait une préférence de place conditionnelle. Ainsi, la signalisation des endorphines dans le gyrus cingulaire antérieur, une zone de prise de décision et de rechute, est importante pour l’analgésie.
Un autre domaine de recherche concerne les neurones de l’habénula latérale (LHb) connus pour être en corrélation avec des états aversifs tels que la douleur, l’abstinence aux opiacés, les modèles de dépression chez les rongeurs et le fait de ne pas recevoir une récompense prévue. Margolis et Fields [64] ont découvert que l’agoniste opioïde DAMGO inhibe paradoxalement la libération de glutamate et de GABA chez les rongeurs. Ils suggèrent que les effets comportementaux de l’activation du MOR dépendent de l’activité neuronale intrinsèque de la LHb et de l’équilibre de l’activité des entrées de glutamate et de GABA pour prédire la réponse à la douleur. Les recherches menées dans le cadre d’expériences psychopharmacologiques sur les opioïdes dans des modèles animaux précliniques fournissent plusieurs phénotypes comportementaux induits observés dans l’OUD humaine, notamment la vulnérabilité aux facteurs de stress [65], la diminution de la motivation pour les récompenses naturelles [66], la réduction des tâches d’apprentissage et de mémoire [67], le retrait social [68], les symptômes de sevrage [69], l’anxiété et les comportements de type dépressif [70].
En outre, l’excitabilité des neurones glutamatergiques qui se projettent dans l’accumbens peut réguler la consommation et la recherche d’opioïdes [71]. Plus précisément, un traitement chronique à la morphine augmente la fréquence de déclenchement des afférences glutamatergiques provenant de l’amygdale, mais pas du PFC. Pour mieux comprendre le rôle de l’activité glutaminergique dans l’OUD, des études ont révélé qu’il existe deux sous-types principaux de neurones à épines moyennes (MSN) dans le NAc qui expriment à la fois des récepteurs D1 et D2 de la dopamine [72]. Une analyse minutieuse de ces études et d’autres [73,74] révèle que la plasticité de la transmission excitatrice au sein du NAc sur les MSN D1 est liée à une recherche accrue d’opioïdes [60]. En revanche, une plasticité et une signalisation similaires au niveau des D2-MSN déclenchent des comportements liés à l’extinction [75]. Il s’ensuit donc qu’à l’intérieur de l’enveloppe du NAc, une libération accrue de glutamate peut se produire au niveau des synapses D1-MSN, tandis qu’une libération atténuée de glutamate peut se produire au niveau des synapses D2-MSN après une exposition chronique à la morphine [76].
En outre, le retrait de la morphine augmente l’expression de l’ARNm c-Fos dans les projections thalamiques paraventriculaires (PVT) vers la coquille médiane du NAc [77], spécifique des D2-MSN. Cependant, chez les souris transgéniques MOR nulles, la réintégration de MOR dans les MSN D1 du striatum rétablit la perte de la préférence de place conditionnée induite par les opioïdes (morphine) [78]. Sur la base de ces recherches et d’autres [78], l’activation de la commande glutaminergique dans la VTA par l’administration de N-acétylcystéine semble être une cible focale pour aider à réguler la libération de dopamine dans le RDS [79,80]. Étant donné que le déficit endorphinergique a un impact sur le RDS, l’utilisation potentielle d’inhibiteurs de l’enképhalinase pour augmenter les endorphines dans les régions cérébrales mésolimbiques et PFC semble raisonnable. Chez des souris C57J génétiquement sélectionnées et aimant l’alcool, ayant un faible niveau de méthionine enképhaline dans le cerveau et comparées à des souris DBA, l’inhibition de l’enképhalinase avec de la D-phénylalanine a entraîné une augmentation des niveaux d’endorphines dans l’hypophyse et le striatum avec une réduction concomitante de la consommation d’alcool [80].
Une nouvelle approche thérapeutique, “Precision Addiction Management” (gestion précise de la dépendance)
Les corrélats neurologiques responsables du RDS identifiés par le test GARS sont utilisés pour personnaliser la régulation de la pro-dopamine en fonction des résultats du test. Le régulateur de pro-dopamine personnalisé est un neutraceutique composé d’une inhibition de l’enképhalinase, d’acides aminés précurseurs et d’une inhibition du catabolisme avec le code d’identification de recherche KB220. Au cours des cinquante années de développement des variantes nutraceutiques du KB220, au moins 42 études ont été publiées, y compris des modèles animaux d’addiction, des essais cliniques humains, des études en double et triple aveugle contrôlées par placebo et des études de neuro-imagerie [79]. Le nombre d’études sur chaque ingrédient du KB220 trouvées dans PubMed le 12 janvier 2020 est listé dans le tableau 3.
Études futures
Des recherches supplémentaires sont encouragées. L’une d’entre elles consiste à étudier le trait polymorphe de l’allèle Taq A1 du récepteur D2 de la dopamine, connu pour coder pour environ 30 % de récepteurs DRD2 en moins, et à évaluer le traitement avant et après 30 jours avec des variantes du KB220. La proposition est qu’après le KB220, il y aura une augmentation des récepteurs DRD2 mesurée par l’expression de l’ARNm.
Bien que nous ayons déjà montré que l’agent pro-dopaminergique KB220Z produisait une augmentation du volume et de la connectivité fonctionnelle entre les centres de récompense et les centres cognitifs du cerveau de rongeurs naïfs [81] ainsi qu’une augmentation similaire de la connectivité fonctionnelle et du volume entre les centres cognitifs et les centres de récompense de sujets abstinents dépendants de l’héroïne [82], nous encourageons la communauté neuroscientifique à mener une série d’autres études croisées contrôlées par placebo pour aider à établir que le KB220 est un agoniste dopaminergique.
En ce qui concerne les études futures visant à fournir des données démontrant que le KB220 a des effets homéostatiques ou psychopharmacologiques dopaminergiques, nous proposons ce qui suit : 1. Des études de microdialyse pour montrer l’effet de l’administration aiguë de KB220 sur la libération de DA par la NAC [83] ; 2. Montrer que si l’administration de KB220 réduit la disponibilité des récepteurs D2 et D3, cette réduction pourrait indiquer une meilleure occupation des récepteurs dopaminergiques par rapport au placebo [84]. 3. Expériences d’auto-administration avec le KB220 comparé au placebo, y compris l’acquisition de l’auto-administration du KB220, l’extinction et les tests de réintégration, la réintégration induite par le stress, l’administration intracrânienne du KB220 sur la réintégration induite par le stress [85]. 4. Localisation de l’ARNm du récepteur D2 de la dopamine dans le cerveau du rat par hybridation in situ histochimique après administration aiguë et chronique par rapport au placebo [86] et 5. Utilisation de la TEP pour suivre des sujets porteurs de l’allèle DRD2 A1, par rapport à l’allèle DRD2 A2, avant et après un traitement chronique (au moins 30 jours) au KB220, pour une augmentation potentielle du nombre de récepteurs DRD2 [87].
Résumé
Il est bien connu que les opioïdes exogènes provoquent le syndrome de déficit en dopamine (SDD), un sous-ensemble du SDR, ainsi que l’anhédonie et la recherche et l’utilisation d’opioïdes exogènes. L’un des principaux problèmes thérapeutiques actuels est la volonté de nombreux cliniciens d’accroître l’utilisation des MAT, en particulier de la buprénorphine, pour traiter le SDD et le SDO en tant que traitement de substitution à vie. Nous avons soutenu que ce type de traitement, qui substitue un narcotique à un autre narcotique, est apparemment contre-intuitif et qu’il peut réduire les dommages sociétaux, mais qu’il perpétuera également la dépendance aux opioïdes.
Par conséquent, dans cet article, nous proposons une “gestion de précision de la dépendance” à la suite des recherches nécessaires ; le traitement de première ligne devrait consister en un test GARS pour diriger la restauration de précision des neurotransmetteurs polymorphes.
