Comer, S. D., & Cahill, C. M. (2019). Fentanyl: Receptor pharmacology, abuse potential, and implications for treatment. Neuroscience & Biobehavioral Reviews, 106, 49-57.
Abstract
Les surdoses d’opioïdes, dont beaucoup sont attribuées à l’utilisation de fentanyl illicite, sont actuellement l’une des principales causes de décès aux États-Unis. Bien que le fentanyl ait été utilisé en toute sécurité pendant des décennies dans des contextes cliniques, l’utilisation généralisée de fentanyl illicite est un phénomène récent. À partir de 2013, le fentanyl et ses analogues fabriqués illicitement ont commencé à apparaître dans les rues. Ces substances étaient ajoutées ou vendues comme de l’héroïne, souvent à l’insu de l’utilisateur. Le fentanyl étant très puissant, seules de petites quantités sont nécessaires pour produire des effets pharmacologiques, mais la marge entre les doses sûres et les doses toxiques est étroite. On sait étonnamment peu de choses sur les mécanismes de signalisation exacts qui sous-tendent la dépression respiratoire liée au fentanyl ou sur l’efficacité de la naloxone pour inverser cet effet. De même, on sait peu de choses sur la capacité des médicaments de traitement tels que la buprénorphine, la méthadone ou la naltrexone à réduire la consommation de fentanyl illicite. Le présent article passe en revue le récepteur, la pharmacologie préclinique et clinique du fentanyl et la façon dont sa pharmacologie peut prédire l’efficacité des médicaments actuellement approuvés pour traiter l’usage illicite de fentanyl.
1. Introduction : développement du fentanyl
Paul Janssen a synthétisé le fentanyl en 1960 en partant du principe que la synthèse d’un médicament très puissant avec une spécificité accrue des récepteurs présenterait un profil de sécurité supérieur à celui de la morphine. Au départ, le médicament a été approuvé aux États-Unis uniquement en association avec le dropéridol en raison des inquiétudes suscitées par son extrême puissance et sa plus grande propension à produire une rigidité musculaire par rapport à d’autres opioïdes. Malgré ces préoccupations initiales, la capacité du fentanyl à assurer la stabilité cardiovasculaire et à bloquer la réaction de stress aux stimuli chirurgicaux à des doses élevées en a fait le pilier de l’anesthésie cardiaque. L’utilisation clinique du fentanyl a été limitée à l’anesthésie jusqu’aux années 1990, lorsque le développement de formulations non injectables a été poursuivi. Aujourd’hui, de nombreux produits à base de fentanyl seul sont autorisés aux États-Unis, notamment des pastilles transmuqueuses orales, des comprimés buccaux effervescents, des comprimés sublinguaux, des sprays sublinguaux, des sprays nasaux, des timbres transdermiques et des formulations injectables. Ces produits sont utilisés comme agents anesthésiques en chirurgie, pour le traitement de la douleur chronique et comme médicaments d’appoint en cas d’accès de douleur chez les patients atteints de cancer. Un certain nombre d’autres médicaments dont la structure chimique est similaire à celle du fentanyl ont été synthétisés (par exemple, le sufentanil, l’alfentanil et le carfentanil), dont l’utilisation en anesthésie clinique est restreinte ou plus rarement utilisée pour les blocs nerveux ou, dans le cas du carfentanil, comme radiotraceur dans les études de recherche utilisant la tomographie par émission de positrons (TEP). Ces médicaments sont nettement plus puissants que la morphine : par exemple, le carfentanil est 10 000 fois plus puissant que la morphine en tant qu’analgésique.
2. Épidémiologie de l’utilisation illicite du fentanyl
Malgré l’utilisation courante du fentanyl en milieu clinique, une autre préoccupation qui a retardé son approbation initiale aux États-Unis était son potentiel d’abus. La Drug Enforcement Agency des États-Unis a finalement placé le fentanyl, ainsi que les autres médicaments de type fentanyl, notamment le sufentanil, l’alfentanil et le carfentanil, dans l’annexe II de la loi sur les substances contrôlées (Controlled Substances Act) parce qu’elle estimait qu’ils présentaient un fort potentiel d’abus. Pendant les décennies qui ont suivi l’approbation du fentanyl, les rapports faisant état d’abus ont toutefois été peu nombreux par rapport à d’autres produits opioïdes délivrés sur ordonnance, tels que l’oxycodone et l’hydrocodone. La plupart des premiers rapports suggéraient que le fentanyl était consommé par des professionnels de la santé, comme les anesthésistes, qui y avaient facilement accès. Bien que des rapports ultérieurs aient décrit l’utilisation non médicale du dispositif transdermique de fentanyl par des patients et/ou des personnes souffrant de troubles liés à l’utilisation de substances, les taux de prévalence globaux de l’utilisation non médicale de produits à base de fentanyl approuvés par la FDA sont restés faibles. Cependant, en 2006, les États-Unis ont connu une augmentation des décès par overdose liés au fentanyl et des saisies de fentanyl fabriqué illicitement par la Drug Enforcement Agency (DEA). Cette “crise” a été attribuée au mélange de fentanyl avec de l’héroïne. L’origine de cette crise a été attribuée à un seul laboratoire clandestin qui fabriquait du fentanyl de manière illicite. Lorsque ce laboratoire a été fermé, le nombre de décès par overdose de fentanyl et les saisies de fentanyl effectuées par la DEA ont rapidement diminué.
Dans la crise actuelle du fentanyl, de nombreux laboratoires clandestins dans le monde fabriquent du fentanyl illicite ainsi qu’un certain nombre d’autres composés ayant des structures chimiques similaires qui, jusqu’à très récemment, auraient échappé à la programmation de la DEA, mais qui sont désormais couverts par une loi dérivée pour éviter d’échapper aux poursuites. À partir de 2013, les saisies de fentanyl ont augmenté de façon spectaculaire aux États-Unis et, en 2015, le nombre de saisies de fentanyl était environ huit fois plus élevé qu’en 2006. La synthèse du fentanyl est relativement simple par rapport à celle de l’héroïne et, comme il est très puissant, il est facile de le dissimuler et de le transporter pour le vendre, ce qui réduit les risques de détection et d’arrestation pour les trafiquants de drogue. Les dealers l’achètent à bas prix et l’ajoutent à l’héroïne à l’insu de l’usager, ce qui leur permet de réaliser d’énormes profits. En plus d’être utilisé comme adultérant de l’héroïne, le fentanyl est vendu sous forme de pilules comme du faux Norco®, un médicament contre la douleur délivré sur ordonnance contenant de l’hydrocodone et de l’acétaminophène, ou CDN 80, qui est censé imiter un médicament contre la douleur délivré sur ordonnance contenant de l’oxycodone et vendu au Canada. Le fait que du fentanyl soit ajouté à la cocaïne et vendu sous forme de pilules de Xanax® contrefaites (un anxiolytique à base de benzodiazépine à courte durée d’action utilisé pour traiter les troubles anxieux) est tout aussi préoccupant, voire plus. Comme les consommateurs de ces substances ont généralement peu ou pas de tolérance aux opioïdes, le risque d’overdose peut être plus élevé. L’augmentation considérable de la disponibilité du fentanyl illicite a été associée à une hausse des décès par overdose. Plus de 63 000 Américains sont morts d’overdoses en 2016, dont plus de 19 000 étaient liés à des opioïdes synthétiques tels que le fentanyl et ses analogues. Il est à craindre que le nombre d’overdoses et de décès dus au fentanyl continue d’augmenter dans les années à venir. Malgré ces tendances alarmantes, on en sait relativement peu sur les mécanismes de signalisation exacts qui contribuent aux surdoses et aux décès liés au fentanyl, et sur l’efficacité des médicaments actuels approuvés par la FDA pour le traitement des troubles liés à l’utilisation d’opioïdes contre le fentanyl. Les sections suivantes de cette revue décriront la pharmacologie des récepteurs du fentanyl, les données précliniques sur son risque d’abus, la pharmacologie clinique du fentanyl en relation avec le risque d’abus, et leurs implications pour le traitement de l’abus de fentanyl.
3. Pharmacologie du fentanyl
Comme la plupart des opioïdes utilisés en clinique, le fentanyl produit ses effets pharmacologiques via l’activation du récepteur opioïde mu (MOR) avec une faible affinité pour les récepteurs opioïdes delta et kappa. Le fentanyl est un agoniste opioïde phénylpipéridine synthétique et lipophile, contrairement à la morphine, qui est un alcaloïde extrait du pavot à opium. Le fentanyl est un agoniste très efficace au niveau du MOR avec une affinité de liaison (Ki) de 1,35 nM au niveau des MOR humains recombinants, une affinité similaire à celle rapportée en utilisant des membranes de cobaye (1,2 nM Ki). Cependant, une large gamme d’affinités de liaison du fentanyl pour le MOR a été rapportée (Ki = 0,007 à >200 nM), ce qui reflète très probablement des différences dans le radioligand, l’espèce, l’essai ou le tissu utilisé. Cette affinité est très similaire à la liaison de la morphine au MOR (Ki = 1,17 nM). En outre, la demi-vie d’élimination est similaire entre le fentanyl et la morphine avec des t1/2 de 2-4 h pour le fentanyl et de 2 h pour la morphine. Cela peut être surprenant, étant donné que le fentanyl a un début d’action plus rapide, une durée d’action analgésique beaucoup plus courte et une puissance analgésique plus élevée que celle de la morphine. Des études humaines et précliniques montrent que le fentanyl est 50 fois (intramusculaire), 150 fois (sous-cutané), ∼400 fois (intraveineux) ou 10 fois (épidural) plus puissant que la morphine, mais la plupart des médecins acceptent et les tableaux de conversion indiquent que le fentanyl est environ 100 fois plus puissant que la morphine. En outre, le fentanyl traverse rapidement la barrière hémato-encéphalique, ce qui lui confère un pouvoir analgésique plus important, qui se traduit par une demi-vie de ∼5 min pour l’équilibre entre le plasma et le liquide céphalo-rachidien. Ainsi, la plus grande puissance analgésique et l’action plus rapide du fentanyl par rapport à la morphine ne s’expliquent pas par l’affinité de liaison ou la demi-vie. Les niveaux de fentanyl diminuent rapidement en raison de la redistribution vers d’autres tissus et le fentanyl est rapidement séquestré dans la graisse corporelle, ce qui contribue à sa courte durée d’action. La différence de puissance, de début et de durée d’action est en partie attribuée à la différence de lipophilie de ces médicaments. Parmi les agonistes MOR disponibles en clinique, le fentanyl et le sufentanil sont les plus liposolubles, alors que la morphine est plus hydrophile. En utilisant un coefficient de partage octanol-eau classique pour mesurer la liposolubilité, le coefficient pour la morphine est de 6 mais >700 pour le fentanyl. La différence de liposolubilité a un impact non seulement sur la voie d’administration pour l’utilisation clinique, mais aussi sur la pharmacocinétique du métabolisme et de l’élimination. En outre, les propriétés pharmacocinétiques du fentanyl ont permis le développement d’indications cliniques uniques de formulations non injectables, allant du traitement de la douleur cancéreuse à l’aide de formulations nasales avec accès direct au cerveau à la libération transdermique pour le traitement de la douleur chronique.
Le fentanyl est faiblement absorbé par le tractus gastro-intestinal, mais il est exclusivement métabolisé, l’excrétion rénale représentant moins de 10 % de la dose. Le métabolisme par N-désalkylation de la pipéridine en norfentanyl, un métabolite inactif, est la voie de dégradation prédominante chez l’homme, représentant 99 % du métabolisme du fentanyl. Le métabolisme du fentanyl est assuré presque exclusivement par le cytochrome P450 CYP3A4, ainsi que par les CYP3A5 et CYP3A7. L’implication du métabolisme dépendant du CYP3A explique de nombreuses interactions médicamenteuses indésirables, notamment avec l’inhibiteur de la protéase du VIH, le ritonavir. On a signalé que le ritonavir et le diltiazem, un inhibiteur des canaux calciques, augmentaient les concentrations plasmatiques et réduisaient l’élimination du fentanyl. Inversement, le fentanyl peut agir comme un inhibiteur enzymatique et réduire la clairance de médicaments sédatifs tels que le midazolam. La courte durée d’action est en partie due à l’activité des glycoprotéines P au sein de la barrière hémato-encéphalique qui pompent le fentanyl hors du système nerveux central (CNS). L’importance de ces protéines est évidente dans la mesure où le lopéramide, un opioïde utilisé pour le traitement de la diarrhée, a des effets négligeables sur le SNC, mais cette restriction périphérique est uniquement due à sa forte affinité pour le substrat P-glycoprotéine ; le lopéramide produit des effets sur le SNC chez les rongeurs knock-out pour la P-glycoprotéine. Les polymorphismes génétiques du gène ABCB1 qui code pour les glycoprotéines P (ABCB1 1236 T T (rs1128503), 2677 T T (rs2032582) et 3435 T T (rs1045642)) entraînent une rétention du fentanyl dans le CNS, ce qui se traduit par des effets indésirables tels que la dépression respiratoire et la sédation. La capacité des opioïdes à produire des effets différents sur la nociception, la dépression respiratoire et la constipation résulte probablement d’une combinaison de leur chimie, qui affecte leur distribution dans le système nerveux central, leur métabolisme, la sélectivité des récepteurs et la signalisation des récepteurs.
4. Pharmacologie préclinique du fentanyl
Les MOR appartiennent à la superfamille des récepteurs couplés aux protéines G, une classe de récepteurs membranaires qui présentent un domaine hélicoïdal à sept transmembranes relié par des boucles intra- et extracellulaires. Les MOR produisent leurs effets par le biais d’interactions avec des protéines G hétérotrimériques inhibitrices (Gi/o), qui sont responsables de la plupart des effets pharmacologiques liés aux opioïdes, notamment l’analgésie et l’euphorie. Cependant, les MOR produisent également une signalisation indépendante de la protéine G par l’intermédiaire des complexes bêta-arrestines. Un effort concerté est actuellement en cours pour identifier des ligands qui favorisent la signalisation par la protéine G avec une moindre activation des bêta-arrestines, car il a été proposé que la signalisation par les arrestines explique les effets dépressifs respiratoires des opioïdes, qui mettent la vie en danger (Fig. 1A). Un agoniste biaisé est défini par la capacité de l’agoniste se liant au même récepteur à activer de manière différentielle les cascades de signalisation, ce qui entraîne la formation de différents complexes protéiques qui déclenchent différents événements cellulaires en aval. La souris knock-out bêta-arrestin-2 est protégée contre la dépression respiratoire et la constipation aiguë induites par la morphine, bien que les effets analgésiques soient renforcés par l’absence de bêta-arrestin 2. Un spectre de biais de signalisation pour différents médicaments opioïdes a été récemment examiné. Le fentanyl présente un biais de signalisation avec une plus grande arrestine par rapport à la signalisation de la protéine G, comme mesuré par la liaison GTPγS dans les cellules exprimant des MOR de souris ou humaines. Cet effet est évident dans les neurones striataux, où l’administration aiguë de fentanyl active la protéine kinase activée par les mitogènes (MAP kinase) ERK1/2 de manière dépendante de la bêta-arrestine, un effet qui est absent après la morphine aiguë, bien que l’activation ERK1/2 soit produite après la morphine chronique. Dans les expériences de culture cellulaire, le fentanyl favorise une phosphorylation robuste du récepteur, le recrutement de la bêta-arrestin-2 et l’internalisation du récepteur, alors que la morphine a des effets beaucoup plus faibles sur ces paramètres. Il est peut-être surprenant, compte tenu du biais agoniste du fentanyl pour la signalisation de la bêta-arrestine, que la tolérance analgésique au fentanyl ne soit pas perturbée chez les souris knock-out pour la bêta-arrestine-2, alors que la tolérance à la morphine est atténuée. La discussion ci-dessus porte spécifiquement sur les différences entre le fentanyl et la morphine, mais des examens complets des mécanismes qui sous-tendent le développement et le maintien de la tolérance aux opioïdes ont été publiés précédemment.
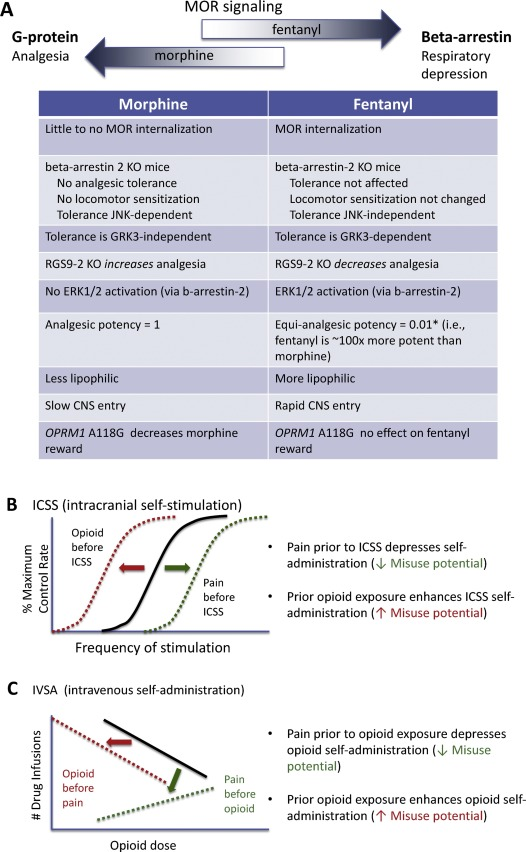
Le fentanyl est un agoniste MOR très efficace qui entraîne une tolérance analgésique moindre que les agonistes MOR moins efficaces tels que la morphine, bien qu’une plus grande tolérance analgésique au fentanyl ait été rapportée après une administration chronique dans des modèles de douleur chronique. Le fentanyl produit une tolérance à court terme (mesurée à l’aide d’un test thermique phasique) par le biais d’un mécanisme dépendant de la kinase du récepteur de la protéine G (GRK3), alors que la morphine produit une tolérance par le biais d’un mécanisme dépendant de la kinase c-Jun N-terminale et non de la GRK3. De même, la signalisation de la cascade JNK dépendant de la bêta-arrestine-2 était responsable de la tolérance analgésique à la morphine et de la sensibilisation locomotrice, mais pas du fentanyl. De nombreuses études ont démontré que le fentanyl et la morphine diffèrent en ce qui concerne les mécanismes de tolérance aux opioïdes et les effets de renforcement. Par exemple, RGS9-2, un régulateur de la signalisation des protéines G, se lie à la sous-unité Gα activée des protéines G, contrôlant ainsi la transduction du signal MOR, la désensibilisation, la tolérance analgésique et la dépendance physique. L’inhibition de la protéine RGS9-2 diminue l’analgésie aiguë induite par le fentanyl mais augmente celle induite par la morphine. En outre, un polymorphisme MOR OPRM1 A118 G (utilisant un modèle de souris humanisé) réduit la capacité de la morphine à potentialiser l’auto-administration intracrânienne (ICSS, un comportement opérant renforcé positivement dans lequel la réponse à la pression du levier est maintenue par l’administration d’une stimulation cérébrale électrique et est une caractéristique du potentiel d’abus) du faisceau cérébral antérieur médian et déprime la libération de dopamine induite par la morphine (impliquée dans la récompense). Toutefois, ce polymorphisme n’a aucun effet sur le SSCI induit par le fentanyl ou sur la libération de dopamine. Une autre mutation ponctuelle T394 A au niveau du site de phosphorylation MOR T394 a diminué la tolérance aux analgésiques opioïdes, mais a augmenté l’auto-administration d’héroïne par voie intraveineuse et la libération de dopamine dans le noyau accumbens, ce qui suggère que cette mutation pourrait augmenter la sensibilité à l’abus d’opioïdes.
L’influence de la douleur sur le potentiel d’abus des analgésiques opioïdes fait encore l’objet d’un grand débat. Dans les modèles de douleur, on pense qu’une dépression de l’ICSS capture la dimension affective de la douleur. Contrairement à un modèle de douleur neuropathique chronique, la douleur viscérale aiguë induite par une injection intrapéritonéale d’acide lactique a déprimé l’ICSS. L’injection systémique d’un agoniste à haute efficacité tel que le fentanyl était plus puissante pour bloquer la dépression de l’ICSS causée par un stimulus douloureux aigu. Dans un modèle de douleur neuropathique chronique, le fentanyl, la méthadone et l’hydromorphone étaient moins puissants pour faciliter l’ICSS (lorsque la stimulation électrique se faisait dans l’aire tegmentale ventrale) par rapport à des témoins naïfs de douleur, ce qui a été interprété comme reflétant un potentiel d’abus réduit des opioïdes dans les états de douleur chronique (Fig. 1B). Une étude antérieure a fait état de résultats similaires pour l’héroïne. Il est intéressant de noter que la morphine n’a pas réussi à faciliter l’ICSS en présence de douleur neuropathique chronique ou dans un modèle de douleur viscérale aiguë.
L’auto-administration de drogues est plus couramment utilisée pour mesurer les effets renforçants des drogues. Des études séminales examinant l’influence de la douleur chronique sur l’auto-administration d’opioïdes ont montré que l’acquisition de l’héroïne, de la morphine, du fentanyl, de l’hydromorphone et de la méthadone par auto-administration était significativement réduite en présence de douleur chronique par rapport à la chirurgie de contrôle sham, mais qu’il n’y avait pas d’effet sur la réponse à la nourriture (Fig. 1C). Il est important de noter que le taux de prise de médicaments est en corrélation avec l’inversion de l’allodynie mécanique. Ces données sont cohérentes avec les rapports selon lesquels la douleur inflammatoire chronique réduit l’acquisition de l’auto-administration de morphine intraveineuse. De même, l’auto-administration de fentanyl par voie orale était réduite ou absente dans trois modèles murins de douleur chronique induite par une douleur inflammatoire à l’adjuvant complet de Freund, une ligature du nerf spinal pour une douleur neuropathique ou une neuropathie induite par la vincristine. À l’appui de l’hypothèse selon laquelle la douleur influence négativement le potentiel d’abus des analgésiques opioïdes, des rapports indiquent que l’administration non contingente d’analgésiques tels que l’indométhacine ou la dexaméthasone a diminué l’auto-administration de morphine par voie intraveineuse ou de fentanyl par voie orale, respectivement. Cependant, d’autres ont rapporté une augmentation de la consommation de fentanyl par voie orale dans un modèle de polyarthrite par rapport à une douleur non douloureuse ou à une douleur neuropathique chronique chez le rat. En revanche, la consommation d’héroïne par voie intraveineuse augmentait si la douleur était induite après que les rongeurs étaient déjà dépendants des opioïdes, ce qui est cohérent avec les études cliniques montrant que les patients souffrant de douleurs chroniques présentaient un risque accru de mésusage d’analgésiques opioïdes s’ils avaient des antécédents d’abus de substances, une consommation actuelle élevée d’alcool, une utilisation à long terme de benzodiazépines ou un comportement aberrant lié à la drogue. Dans des conditions non douloureuses, les rongeurs et les primates non humains ayant un accès prolongé à l’auto-administration d’héroïne par voie intraveineuse augmentent rapidement leur consommation de drogue, ce qui va de pair avec le développement d’une tolérance aux analgésiques et la poursuite de la consommation de drogue malgré des conséquences néfastes telles qu’un choc au pied.
5. Pharmacologie clinique du fentanyl : accent sur le potentiel d’abus
5.1 Volontaires sains
Certaines des premières études sur le risque d’abus du fentanyl ont été menées sur des volontaires sains et normaux qui ne consommaient pas de drogues à des fins récréatives, bien que la plupart des participants consommaient de l’alcool de temps en temps. Dans cette population, le fentanyl n’a pas augmenté de façon fiable les réponses subjectives positives. À des doses intraveineuses (i.v.) allant jusqu’à environ 250 μg/70 kg, le fentanyl a augmenté les effets subjectifs positifs dans 4 études. Dans 3 autres études évaluant des doses de fentanyl i.v. comprises entre 50 μg/70 kg et environ 200 μg/70 kg, le fentanyl n’a pas augmenté les réponses subjectives positives, et dans 2 autres études, les effets subjectifs positifs produits par le fentanyl étaient équivoques. Les résultats négatifs rapportés par Ghoneim et al. (1975) et Scamman et al. (1984) étaient peut-être dus au fait que les effets maximaux du fentanyl n’ont pas été observés parce que les mesures des réponses subjectives n’ont pas commencé avant 30 minutes après l’administration du médicament. Zacny et al. (1996a) ont rapporté que les participants ont déclaré se sentir “high” et “coasting (spaced out)” après avoir reçu 100 μg/70 kg de fentanyl par voie i.v., mais les évaluations de l’appréciation du médicament n’étaient pas significativement différentes de celles de la solution saline par voie i.v. Dans l’une des deux études dont les résultats sont équivoques, l’appréciation de la drogue était transitoire et ne coïncidait pas avec une augmentation des scores sur l’échelle du groupe morphine-benzédrine (MBG) de l’Addiction Research Center Inventory (ARCI), une mesure largement utilisée à l’époque pour évaluer l’euphorie induite par la drogue. Dans l’autre étude rapportant des résultats équivoques, seul un sous-ensemble de participants (4 sur 6) a déclaré aimer les effets du fentanyl, mais une dose relativement faible a été testée (50 μg/70 kg i.v.), de sorte que ce résultat n’est peut-être pas tout à fait surprenant.
5.2 Consommateurs d’opioïdes illicites – effets subjectifs
Selon les normes actuelles, la plupart des évaluations du risque d’abus de drogues sont menées auprès d’individus qui en font un usage récréatif. Il est généralement admis que les consommateurs de drogues à usage récréatif constituent la population la plus appropriée pour tester le risque d’abus de drogues car, par leur comportement, ces personnes ont démontré qu’elles pouvaient reconnaître les effets des drogues et qu’elles les aimaient, généralement à des doses plus élevées que celles utilisées à des fins thérapeutiques. En 2017, la Food and Drug Administration (FDA) a publié un document d’orientation à l’intention de l’industrie qui recommandait que les consommateurs de drogues à usage récréatif ayant des antécédents récents de consommation de substances appartenant à la même classe de drogues que le composé à tester soient recrutés pour évaluer le risque d’abus de drogues. La FDA précise dans son document d’orientation qu'”il n’est pas recommandé d’utiliser des sujets n’ayant jamais consommé de drogues dans les études HAP [human abuse potential] car cette population n’a pas été validée scientifiquement comme étant capable de fournir des informations précises sur le potentiel d’abus d’un médicament”.
Cette recommandation est étayée par le fait que toutes les études portant sur les effets subjectifs du fentanyl chez des consommateurs de drogues expérimentés ont montré que le fentanyl produit des augmentations claires et liées à la dose des évaluations de l’appréciation de la drogue, des bons effets de la drogue et de l’état d’euphorie. Des participants maintenus sous morphine (30 mg par voie orale, administrés 4 fois par jour) ont déclaré qu’ils paieraient 8,50 $ pour une dose de fentanyl de 250 μg/70 kg i.v., contre 2,50 $ pour une solution saline, et les évaluations des “mauvais effets de la drogue” et des “nausées” n’étaient pas significativement différentes de celles de la solution saline. Conformément à ces résultats, des doses allant jusqu’à 4,5 μg/kg (∼315 μg/70 kg) n’ont pas augmenté de manière significative l’évaluation des “mauvais effets” ou de la “maladie” chez des utilisateurs récréatifs d’opioïdes non dépendants (Baylon et al., 2000). En revanche, 7 des 8 volontaires sains ayant reçu 3 μg/kg (∼210 μg/70 kg) ont eu des nausées et 4 d’entre eux ont vomi ; 3 des 4 personnes ayant vomi l’ont fait jusqu’à 6 heures après l’administration du fentanyl. Des vertiges ont été signalés par un autre sujet, qui est resté couché pendant 8 heures après l’administration du médicament. Il n’est pas surprenant que des personnes inexpérimentées en matière de drogues ne déclarent pas aimer les effets du fentanyl.
5.3 Consommateurs d’opioïdes illicites – effets de renforcement
Outre l’évaluation des réponses subjectives après l’administration d’une drogue, le potentiel d’abus des drogues chez l’homme peut être évalué par des procédures d’auto-administration. En règle générale, les participants sont invités à donner une réponse (par exemple, appuyer avec le doigt sur une souris d’ordinateur) pour obtenir la drogue, et une drogue qui est auto-administrée plus souvent qu’un placebo est considérée comme un renforçateur. Une procédure d’évaluation des effets renforçants d’une drogue utilise un schéma modifié de rapport progressif entre la drogue et l’argent pour évaluer les effets renforçants. Les participants reçoivent d’abord un échantillon de drogue et d’argent, puis, au cours d’une session ultérieure, ils ont 10 occasions de choisir entre 1/10e de la dose ou de l’argent qui a été échantillonné précédemment. Chaque fois que la drogue ou l’argent est choisi, le nombre de réponses (pressions du doigt) augmente progressivement et le point où la réponse s’arrête est appelé “point de rupture” (il s’agit du ratio le plus élevé atteint pour la drogue et/ou l’argent). Chez les personnes dépendantes de la morphine, 250 μg/70 kg i.v. de fentanyl ont produit une valeur de point de rupture du rapport progressif pour la drogue qui était significativement supérieure au placebo et similaire à 12,5 mg/70 kg i.v. d’héroïne, 25 mg/70 kg i.v. d’oxycodone et 25 mg/70 kg i.v. de morphine. Le fentanyl a également servi de renforçateur chez des personnes traitées à la méthadone.
Une autre façon d’évaluer l’auto-administration de drogues consiste à utiliser des procédures “économiques comportementales”. Une nouvelle analyse des données a été réalisée à partir d’études utilisant une procédure à choix multiples (dans laquelle les sujets devaient faire une série de choix entre recevoir une dose donnée de médicament et une gamme de montants d’argent). Dans cette analyse économique comportementale de données de procédures à choix multiples, des ” courbes de demande ” ont été construites en traçant les choix de médicaments en fonction du prix unitaire (exigence de réponse divisée par la dose) pour le fentanyl, l’hydromorphone et la méthadone. La courbe de demande de fentanyl était la plus “inélastique” des opioïdes testés, ce qui suggère que l’auto-administration de fentanyl est la plus résistante aux changements lorsque le prix unitaire augmente. Cependant, plusieurs différences de procédure entre les études à partir desquelles l’analyse a été réalisée peuvent expliquer ce résultat, comme les différences de voie et de méthode d’administration du médicament (dosage cumulatif du fentanyl par voie intraveineuse contre dosage aigu de l’hydromorphone par voie intramusculaire). Par conséquent, l’interprétation de l’élasticité du fentanyl par rapport aux autres opioïdes doit être faite avec prudence.
Une autre étude intéressante portant sur les effets de renforcement du fentanyl a été réalisée auprès de consommateurs de drogues récréatives (dont un seul a déclaré consommer des opioïdes à des fins récréatives) à qui l’on a demandé d’immerger leur avant-bras dans de l’eau maintenue à différentes températures (37 °C, 10 °C et 2 °C). Pour chacune des températures, les participants ont effectué deux essais consécutifs d’échantillonnage et trois essais consécutifs de choix. Ils devaient choisir l’une des deux pompes à perfusion (contenant soit du sérum physiologique, soit 50 μg de fentanyl i.v.) cinq minutes avant l’immersion de l’avant-bras dans l’eau. Dans ces conditions, le fentanyl a été auto-administré significativement plus que le placebo dans les deux conditions d’eau froide (77 % du temps dans les conditions de 10 °C et de 2 °C) mais pas lorsque l’eau était maintenue à 37 °C (le fentanyl a été choisi 60 % du temps, ce qui ne différait pas du hasard). La présence de douleur a également modifié les effets subjectifs du fentanyl : les participants ont déclaré se sentir plus excités après l’administration de fentanyl par rapport à la solution saline dans la condition 37 °C, mais pas lorsqu’on leur a demandé d’immerger leur avant-bras dans de l’eau froide (conditions 10 °C et 2 °C). Certains de ces résultats ont été reproduits dans une étude ultérieure : l’oxycodone n’a été auto-administrée qu’en présence d’un stimulus douloureux (immersion des mains dans de l’eau maintenue à 2 °C), par rapport à un stimulus non douloureux (immersion des mains dans de l’eau maintenue à 37 °C). Cependant, ce résultat ne s’est produit que chez les participants qui avaient consommé des opioïdes sur ordonnance à des fins médicales, mais qui n’en avaient jamais consommé à des fins récréatives. Les participants ayant consommé des opioïdes sur ordonnance à des fins récréatives se sont auto-administrés de l’oxycodone, indépendamment de la présence ou de l’absence de douleur (conditions à 4 °C et 37 °C). Et contrairement aux résultats rapportés par Zacny et al. (1996b), les réponses subjectives positives produites par l’oxycodone ne différaient pas en présence et en absence de douleur dans les deux groupes. Ainsi, l’absence d’effets renforçants du fentanyl en l’absence de douleur dans l’étude menée par Zacny et al. (1996b) peut être due au fait que les participants n’étaient pas des utilisateurs récréatifs d’opioïdes.
Les études examinées ci-dessus mettent en évidence plusieurs facteurs importants qui doivent être pris en compte lors de l’évaluation et de l’interprétation des résultats des études sur le potentiel d’abus chez l’homme, notamment la population sélectionnée pour l’étude (les utilisateurs récréatifs d’opioïdes doivent être examinés), les moments d’évaluation utilisés (ils doivent refléter le profil pharmacocinétique attendu du médicament, en particulier aux premiers moments après l’administration du médicament), et l’utilisation de critères comportementaux tels que l’auto-administration du médicament pour fournir plus de clarté sur le risque d’abus d’un médicament. Lorsque tous ces facteurs sont pris en compte, le profil pharmacologique du fentanyl suggère qu’il présente un potentiel élevé d’abus chez l’homme. Cependant, le risque d’abus du fentanyl par rapport à d’autres agonistes opioïdes mu reste assez flou. L’analyse de Greenwald (2008) suggère que le fentanyl pourrait présenter un risque d’abus plus élevé que l’hydromorphone et la méthadone, mais les incohérences procédurales des études examinées rendent difficiles les conclusions définitives. L’étude de Comer et al. (2008) a montré que le fentanyl est plus puissant que l’héroïne, la morphine et l’oxycodone, mais qu’il présente un risque d’abus similaire à celui des autres drogues. Dans cette étude, il aurait été utile de tester des doses plus élevées de fentanyl et d’utiliser des valeurs de ratio progressif plus élevées pour éviter les effets de plafond. De futures études utilisant des mesures potentiellement plus sensibles, telles qu’une procédure de choix entre deux médicaments ou des évaluations prospectives des courbes de demande de fentanyl par rapport à d’autres opioïdes mu, seraient instructives. Une autre façon d’aborder cette question consiste à demander directement aux consommateurs d’opioïdes comment ils perçoivent les effets du fentanyl. Cicero et al. (2017) ont interrogé 10 900 personnes qui commençaient un traitement pour un trouble lié à l’utilisation d’opioïdes sur le fentanyl. Cette analyse a toutefois été entravée par plusieurs variables, notamment le fait que des produits de fentanyl commerciaux et illicites sont disponibles pour les utilisateurs et qu’il est impossible de les distinguer sur la base des dépistages urinaires, que le fentanyl illicite est le plus souvent ajouté à l’héroïne et à d’autres drogues à l’insu de l’utilisateur, et que la mesure dans laquelle le fentanyl illicite seul est disponible pour les utilisateurs et recherché par ces derniers n’est pas claire. Toutefois, compte tenu des modes actuels de fabrication illicite, des techniques modernes de commercialisation et des énormes profits à réaliser, il est probable que la consommation de fentanyl illicite se généralisera encore dans les années à venir.
6. Implications pour le traitement de l’usage illicite de fentanyl
Les données précliniques examinées ci-dessus confirment que la pharmacologie du fentanyl diffère de celle d’autres agonistes opioïdes mu tels que la morphine. En revanche, on ne sait pas si la pharmacologie du fentanyl chez l’homme, en ce qui concerne le risque d’abus, diffère significativement de celle des autres opioïdes mu, en partie parce que les procédures de recherche qui pourraient potentiellement établir cette différenciation (par exemple, un paradigme de choix drogue contre drogue ou des procédures prospectives d’économie comportementale) n’ont pas été appliquées à cette question. La question de savoir si la pharmacologie du fentanyl chez l’homme en ce qui concerne la toxicité diffère de celle des autres opioïdes a également été peu étudiée, même si la toxicité du fentanyl en milieu clinique a été bien caractérisée. S’il est bien connu que le fentanyl, comme d’autres agonistes opioïdes, produit une dépression respiratoire principalement par l’activation des récepteurs opioïdes du complexe de pré-Bötzinger ainsi que par des actions dans les noyaux de Kolliker-Fuse et parabrachial du pons, des études cliniques récentes ont également démontré que le fentanyl induit une rigidité de la paroi thoracique qui peut contribuer à des décès. En outre, la combinaison du fentanyl avec d’autres drogues d’abus ou des dépresseurs du CNS tels que l’alcool engage probablement des mécanismes supplémentaires, y compris des arythmies cardiaques, qui conduisent à la mortalité. Le manque de connaissances sur la manière dont le fentanyl peut différer des autres agonistes opioïdes est principalement dû au fait que le fentanyl est utilisé d’une manière très différente par un clinicien administrant le médicament à un patient par rapport à un toxicomane s’auto-administrant du fentanyl pour ses effets euphorisants (c’est-à-dire une dose bolus importante injectée très rapidement, souvent en combinaison avec de l’alcool ou d’autres drogues d’abus telles que la cocaïne ou les benzodiazépines).
Outre les lacunes de la recherche concernant le risque d’abus et la toxicité du fentanyl par rapport à d’autres agonistes opioïdes, on dispose de peu d’informations provenant d’essais cliniques contrôlés sur l’efficacité des médicaments de traitement (méthadone, buprénorphine, naltrexone) pour réduire l’usage illicite de fentanyl, ou de la naloxone pour traiter les surdoses liées au fentanyl. Des études précliniques ont clairement établi que le fentanyl interagit de manière compétitive avec les antagonistes opioïdes tels que la naltrexone. Ainsi, une simple augmentation de la dose d’antagoniste devrait être efficace si les effets euphoriques du fentanyl ne sont pas complètement supprimés (naltrexone) ou si les effets dépresseurs respiratoires du fentanyl ne sont pas complètement inversés (naloxone). Une mise en garde importante concernant cette dernière affirmation est que l’efficacité de la naloxone pour inverser les surdoses liées au fentanyl n’est pas claire lorsque de l’alcool ou d’autres drogues ont été ingérés en même temps que le fentanyl ou lorsqu’une drogue synthétique semblable au fentanyl a été utilisée.
La naloxone est utilisée depuis des décennies pour inverser la dépression respiratoire induite par les opioïdes, à la fois en milieu hospitalier (par exemple, lors d’une intervention chirurgicale) et en milieu extrahospitalier (par exemple, en cas d’overdose chez un consommateur de drogues illicites). Son action est rapide (dans les 2 minutes suivant l’administration intraveineuse) et sa demi-vie sérique est courte (∼1 h). Chez les personnes dépendantes des opioïdes, la naloxone peut précipiter le sevrage, dont la gravité peut dépendre de multiples facteurs, tels que le niveau de dépendance physique de la personne, la quantité et le type d’agoniste opioïde utilisé lors du surdosage, le temps écoulé entre le surdosage et l’administration de la naloxone, et la quantité de naloxone utilisée pour inverser le surdosage. Afin d’éviter de précipiter un sevrage sévère, l’American Heart Association recommande de commencer par une petite dose de naloxone (0,4 mg par voie intramusculaire ou 2 mg par voie intranasale). Cependant, des rapports récents suggèrent que des doses plus élevées ou des doses répétées de naloxone (en raison de la récurrence de la dépression respiratoire) peuvent être nécessaires pour inverser la dépression respiratoire induite par le fentanyl. La raison pour laquelle des doses plus élevées de naloxone peuvent être nécessaires n’est pas tout à fait claire. Il est possible qu’une forte dose de naloxone soit nécessaire simplement parce qu’une forte dose de fentanyl a été utilisée, qu’un analogue du fentanyl qui n’est pas sensible à la naloxone a été utilisé ou, parce que le début de la respiration induite par le fentanyl est si rapide, que la naloxone a été administrée alors que la personne était déjà décédée. Une autre possibilité est que le fentanyl et la naloxone partagent un transporteur d’influx dans le cerveau et que, lorsque de fortes doses de fentanyl sont utilisées, le transporteur est saturé, de sorte que la naloxone n’est pas en mesure de traverser la barrière hémato-encéphalique. De toute évidence, il est absolument nécessaire de réaliser davantage d’études cliniques pour évaluer l’efficacité de la naloxone à inverser la dépression respiratoire induite par le fentanyl et les médicaments synthétiques de type fentanyl après différentes voies d’administration.
L’efficacité de la buprénorphine ou de la méthadone pour réduire l’abus de fentanyl chez l’homme est également largement inconnue. Des études menées sur des rats ont montré que le maintien de la buprénorphine était moins efficace pour réduire les effets analgésiques des agonistes opioïdes à faible efficacité (morphine) qu’à efficacité plus élevée (étonitazène). Une étude a également été menée sur des singes rhésus pour comparer les effets renforçants de différents agonistes opioïdes en présence et en l’absence de dépendance physique à la morphine. Par le mécanisme de la tolérance croisée, on s’attendrait à un déplacement vers la droite des courbes dose-effet des opioïdes lorsque les animaux sont physiquement dépendants de la morphine, par rapport à l’absence de dépendance. Bien que ce résultat ait été démontré pour la plupart des agonistes testés, le déplacement vers la droite de la courbe dose-effet pour l’agoniste le plus efficace, l’alfentanil, était plus faible que pour les agonistes d’efficacité intermédiaire, la morphine et l’héroïne. En outre, les courbes dose-effet des agonistes de moindre efficacité ont été déplacées vers le bas (buprénorphine) ou vers la droite dans une mesure beaucoup plus importante (nalbuphine) que celles des agonistes de plus grande efficacité. Ce schéma d’effets a été démontré chez plusieurs espèces différentes (rats, souris, singes, pigeons) dans le cadre de plusieurs essais expérimentaux (analgésie, discrimination par la drogue, réponse alimentaire contrôlée par un calendrier, auto-administration). Par conséquent, pour l’entretien à la buprénorphine et aux agonistes opioïdes, la conclusion générale est que les effets des agonistes plus efficaces sont plus difficiles à bloquer que ceux des agonistes moins efficaces. Dans la mesure où ces résultats peuvent être extrapolés à l’homme, les données suggèrent que la méthadone et la buprénorphine pourraient être moins efficaces pour traiter l’abus de fentanyl que pour traiter l’abus d’héroïne.
En résumé, on en sait beaucoup sur la pharmacologie du fentanyl dans les modèles précliniques et lorsqu’il est utilisé en thérapeutique chez l’homme pour l’anesthésie ou l’analgésie. Cependant, des études sont désespérément nécessaires pour élucider les mécanismes physiologiques qui sous-tendent le surdosage au fentanyl, afin que des traitements efficaces puissent être développés pour réduire le risque de décès. De même, des études visant à évaluer les doses d’entretien et les schémas posologiques de naltrexone, de méthadone et de buprénorphine les plus efficaces pour traiter l’abus de fentanyl sont nécessaires de toute urgence pour faire face à la crise de santé publique que représente l’utilisation de fentanyl illicite.
