
Zanos, P., Moaddel, R., Morris, P. J., Riggs, L. M., Highland, J. N., Georgiou, P., … & Gould, T. D. (2018). Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacological reviews, 70(3), 621-660.
Abstract
La kétamine, un mélange racémique composé de (S)- et de (R)-cétamine, est utilisée en clinique depuis 1970. Bien qu’elle soit surtout caractérisée par ses propriétés anesthésiques dissociatives, la kétamine exerce également des actions analgésiques, anti-inflammatoires et antidépressives. Nous présentons un examen complet de ces utilisations thérapeutiques, en mettant l’accent sur la dose de médicament, la voie d’administration et l’évolution temporelle de ces effets. Les effets secondaires dissociatifs, psychotomimétiques, cognitifs et périphériques associés à une exposition de courte durée ou prolongée, ainsi qu’à l’utilisation récréative de la kétamine, sont également abordés. Nous décrivons également la pharmacocinétique de la kétamine, y compris son métabolisme rapide et important en norkétamine, déshydronorkétamine, hydroxykétamine et hydroxynorkétamine (HNK). Alors que les propriétés anesthésiques et analgésiques de la kétamine sont généralement attribuées à l’inhibition directe des récepteurs N-méthyl-D-aspartate induite par la kétamine, d’autres cibles pharmacologiques supposées de moindre affinité de la kétamine comprennent, sans s’y limiter, les récepteurs de l’acide γ-amynobutyrique (GABA), de la dopamine, de la sérotonine, sigma, opioïdes et cholinergiques, ainsi que les canaux sodium et nucléotides cycliques activés par hyperpolarisation, gérés par tension. Nous examinons les preuves de la pertinence de ces cibles de la kétamine et de ses métabolites pour les effets cliniques du médicament. Les métabolites de la kétamine peuvent avoir une pertinence clinique plus large que ce qui avait été envisagé précédemment, étant donné que les métabolites HNK ont une efficacité antidépressive dans les études précliniques. Dans l’ensemble, la déconvolution des cibles pharmacologiques de la kétamine et de ses métabolites fournira des informations essentielles au développement de nouvelles pharmacothérapies qui possèdent les effets cliniques souhaitables de la kétamine, tout en limitant les effets secondaires indésirables.
I. Introduction
La (R,S)-kétamine (ci-après dénommée kétamine) est un dérivé de la phénylcyclohexylamine (poids molaire = 237,73) composé de ses deux énantiomères optiques, la (S)- et la (R)-kétamine. Elle a été commercialisée pour l’usage humain en 1970 comme anesthésique i.v. à action rapide. La kétamine a été dérivée de la phencyclidine (PCP) dans le but d’atténuer les graves effets secondaires psychotomimétiques/psychodysleptiques et le potentiel d’abus de la molécule mère, qui a ensuite été retirée du marché en 1978. Cependant, la kétamine induit toujours des effets dissociatifs et présente un potentiel d’abus, bien que dans une moindre mesure que le PCP. Malgré ces effets secondaires, la kétamine s’est révélée être un médicament intéressant en raison de sa courte demi-vie et de l’absence de dépression respiratoire cliniquement significative. Outre son action anesthésique bien caractérisée chez les adultes, les enfants et les patients obstétricaux, la kétamine possède des effets analgésiques, anti-inflammatoires.
A. Effets thérapeutiques cliniques
1. Anesthésique.
La kétamine induit une anesthésie générale et dissociative chez l’animal et chez l’homme. En outre, la kétamine est également utilisée comme adjuvant aux anesthésiques locaux dans la pratique vétérinaire et chez l’homme.
L’anesthésie dissociative – une forme d’anesthésie sans perte totale de conscience mais caractérisée par la catatonie, la catalepsie et l’amnésie – est obtenue chez l’homme à des doses de kétamine allant de 1 à 2 mg/kg administrées par voie i.v. (bolus) ou de 4 à 11 mg/kg administrées par voie i.m. Des concentrations plasmatiques maximales de kétamine d’environ 1 200 à 2 400 ng/ml, soit 5 à 10 μM, sont nécessaires pour induire une anesthésie dissociative.
La concentration plasmatique moyenne à l’état d’équilibre nécessaire pour obtenir une anesthésie avec la kétamine serait de 2200 ng/ml, soit 9,3 μM. L’administration orale (500 mg) ou intrarectale (8-15 mg/kg) de kétamine est suffisante pour induire une sédation et/ou une anesthésie générale chez l’homme.
Le réveil de l’anesthésie induite par la kétamine se produit à des concentrations plasmatiques allant de 640 à 1100 ng/ml ou 2,7-4,7 μM. White et al. (1985) ont montré que l’administration du mélange racémique de kétamine (perfusion i.v. de 5 à 7 minutes à raison de 50 mg/min pour une dose totale de 275 ± 25 mg) induisait une anesthésie générale chez cinq volontaires adultes en bonne santé, comme l’indiquait l’absence de réflexe paupier. À la fin de la perfusion, il a fallu environ 11 ± 3 minutes aux volontaires pour ouvrir les yeux (1900-3300 ng/ml ou 8,0-14 μM de concentration sérique), et environ 45 ± 10 minutes pour retrouver une bonne orientation de soi, du lieu et du temps (3,78-4,62 μM de concentration sérique).
La (S)-cétamine intranasale à des doses de 3 à 9 mg/kg induit une sédation chez les patients. En tant qu’anesthésique pour l’homme, la (S)-cétamine serait deux fois plus puissante que le mélange racémique et environ trois fois plus puissante que la (R)-cétamine. Plus précisément, la dose i.v. totale nécessaire à l’induction de l’anesthésie est de 275 ± 25 mg pour la kétamine racémique, de 140 ± 21 mg pour la (S)-cétamine et de 429 ± 37 mg pour la (R)-cétamine. Le temps nécessaire pour retrouver une orientation complète de soi, du lieu et du temps après une administration i.v. de 5 à 7 minutes de (S)-cétamine (25 mg/min ; dose totale : 140 ± 21 mg) ou de (R)-cétamine (75 mg/min ; dose totale : 429 ± 37 mg) était de 21 ± 2 minutes (500-900 ng/ml ou 2,1-3,8 μM de concentration sérique) et de 18 ± 3 minutes (2200-3200 ng/ml ou 9,3-13 μM de concentration sérique), respectivement. Ces données indiquent que l’isomère (S)-cétamine est un anesthésique plus puissant que la (R)-cétamine, étant donné qu’une dose trois fois plus élevée de (R)-cétamine est nécessaire pour obtenir un niveau de sédation comparable. En outre, la concentration sérique de (R)-cétamine qui provoque une diminution de la fréquence médiane demi-maximale (IC50) des oscillations électroencéphalographiques a été mesurée à 2000 ± 500 ng/ml (8,0 ± 2,0 µM), contre 1800 ± 500 ng/ml (7,6 ± 2,0 µM) pour le médicament racémique et 800 ± 400 ng/ml (3,4 ± 1,7 µM) pour l’isomère de la (S)-cétamine.
2. Effets analgésiques
Un premier rapport sur les effets analgésiques de la kétamine a été fourni par Weisman (1971), qui a observé ces effets lors d’interventions ophtalmologiques pédiatriques. La kétamine est décrite comme procurant une forme d’analgésie quantitativement et qualitativement similaire aux opioïdes, mais avec moins d’effets dépressifs respiratoires, comme cela a été rapporté chez des patients pédiatriques traités pour des fractures, des brûlures, ou dans des cas d’amputation traumatique. Lorsqu’elle est administrée par voie i.v. ou i.m., les effets analgésiques de la kétamine sont associés à des concentrations plasmatiques comprises entre 70 et 160 ng/ml, soit environ 0,29-0,67 μM.
La kétamine intraveineuse est utilisée comme analgésique pour réduire la douleur postopératoire chronique et aiguë. Une analgésie adéquate est obtenue à des doses subanesthésiques de kétamine, aussi faibles que 0,15-0,25 mg/kg, lorsqu’elle est administrée par voie i.v, ou 0,5-1 mg/kg lorsqu’elle est administrée par voie i.m. à des patients ayant subi un traumatisme aigu. En outre, les effets antinociceptifs et analgésiques de la kétamine ont été observés lorsque la kétamine est administrée de la manière suivante : 1) par voie orale à la dose de 0,5 mg/kg deux fois par jour pendant 15 jours (comme adjuvant à la morphine) ou à la dose unique de 2 mg/kg ; 2) par voie intranasale à une dose allant de 10 à 50 mg deux fois par jour ; 3) par voie transdermique à une dose de 25 mg libérée sur une période de 24 heures ; 4) par voie s.c. à une dose allant de 0,05 à 0,15 mg/kg par heure pendant 7 jours ; et 5) par voie rectale à une dose de 10 mg/kg. Après administration orale, des concentrations sanguines de kétamine plus faibles peuvent être nécessaires pour obtenir une analgésie par rapport aux autres voies d’administration (concentration maximale, Cmax = 45 ± 10 ng/ml ou 0,19 ± 0,04 μM). La perfusion continue d’une dose subanesthésique de kétamine (titrée de 10 à 40 mg/h ; maintenue pendant 5 jours) s’est avérée efficace pour améliorer la douleur chez les patients souffrant du syndrome douloureux régional complexe (SDRC), entraînant des concentrations plasmatiques de (S)- et de (R)-kétamine comprises entre 200 et 225 ng/ml (0,84-0,95 μM).
L’utilisation de la (S)-cétamine intranasale en tant qu’analgésique peut être particulièrement pertinente en milieu préhospitalier, où l’administration i.v. est difficile, et dans les cas où une administration aiguë pour des blessures est nécessaire, car elle réduit les scores de douleur dans les 5 minutes suivant l’administration. À l’instar de leurs effets anesthésiques différentiels, il est prouvé que la (S)-cétamine est un médicament analgésique plus puissant que la kétamine racémique et la (R)-cétamine chez l’homme, bien que la (S)-cétamine produise également plus d’effets secondaires.
3. Antidépresseur
Les preuves de l’action antidépressive de la kétamine remontent aux années 1970. Dans des études précliniques, la kétamine a exercé des effets similaires à ceux observés après l’administration d’antidépresseurs classiques (antidépresseurs tricycliques et inhibiteurs de la monoamine oxydase) chez les rongeurs. En particulier, l’administration orale de kétamine à des souris a inversé l’hyperthermie induite par la réserpine à la dose de 40 mg/kg et a empêché la ptose induite par la tétrabénazine avec une DE50 de 27,6 mg/kg, qui sont des phénotypes inversés par les antidépresseurs classiques. Les premières preuves des propriétés antidépressives possibles de la kétamine chez l’homme ont été décrites en 1973 par Khorramzadeh et Lotfy (1973), qui ont rapporté que la kétamine administrée par voie i.v. à des doses subanesthésiques de 0,2-1,0 mg/kg (bolus i.v.) entraînait une décharge émotionnelle et facilitait la psychothérapie dans une cohorte de 100 patients hospitalisés en psychiatrie. Cependant, les symptômes précis de la dépression qui ont été améliorés par la kétamine n’ont pas été bien définis dans le contexte des critères diagnostiques et des définitions thérapeutiques modernes. Dans cette étude, la kétamine était en fait qualifiée d’agent abréactif général.
La première étude contrôlée par placebo suggérant que la kétamine a des effets antidépresseurs a été rapportée en 2000. D’après les résultats de cette étude, une perfusion i.v. de 40 minutes de 0,5 mg/kg de kétamine a induit une réponse antidépressive robuste et rapide chez des patients souffrant de dépression, par rapport au placebo. Ce résultat a ensuite été reproduit dans un essai clinique randomisé en double aveugle, contrôlé par placebo, impliquant des patients souffrant de dépression majeure réfractaire au traitement. Zarate et al. (2006) ont notamment démontré que la kétamine exerce un effet antidépresseur qui se manifeste dans les deux heures suivant la perfusion et qui dure en moyenne sept jours chez les patients qui n’ont pas répondu à au moins deux antidépresseurs classiques antérieurs. Plusieurs autres essais cliniques ont reproduit ces résultats chez des patients souffrant de dépression réfractaire au traitement. Pour remédier à l’absence d’insu fonctionnel du traitement en raison des effets dissociatifs de la kétamine, qui se produisent même à de faibles doses subanesthésiques, Murrough et al. (2013a) ont utilisé un placebo psychoactif (c.-à-d. le midazolam) et ont démontré un taux de réponse plus élevé chez les patients qui ont reçu de la kétamine (64 %) que chez ceux qui ont reçu du midazolam (28 %). La kétamine exercerait également des effets antidépresseurs chez les patients souffrant de dépression bipolaire (Diazgranados et al., 2010a ; Zarate et al., 2012b). La (S)-cétamine s’est révélée efficace en tant qu’antidépresseur administré par voie i.v. et intranasale (Singh et al., 2016a ; Daly et al., 2018 ; Canuso et al., 2018). D’autres études ont montré que la kétamine réduit les idées suicidaires et diminue l’anhédonie chez les patients souffrant de dépression majeure. La (S)-cétamine intranasale a également réduit les idées suicidaires chez les patients souffrant de dépression.
La dose antidépressive subanesthésique de kétamine la plus couramment utilisée (0,5 mg/kg ; perfusion de 40 minutes) entraîne une concentration plasmatique maximale (Cmax) de ∼185 ng/ml ou ∼0,78 μM de kétamine, calculée à partir des résultats de Zarate et al. (2012a). Néanmoins, il existe des preuves de réponses antidépressives obtenues à des doses aussi faibles que 0,1 mg/kg (perfusion i.v. de 5 minutes ou injection i.m.), entraînant une Cmax de la kétamine de ∼75 ng/ml (0,32 μM-estimé), comme indiqué dans une petite étude pilote (n = 15) croisée en double aveugle, contrôlée par placebo, chez des patients souffrant de dépression résistante au traitement. Bien que cette étude ait indiqué que des doses plus faibles de kétamine, qui produisent moins d’effets secondaires, pourraient être efficaces dans le traitement de la dépression, ce résultat attend d’être reproduit dans une étude de plus grande envergure.
4. Anti-inflammatoire
L’inflammation est un mécanisme homéostatique essentiel utilisé par l’organisme pour lutter contre les infections et guérir les lésions tissulaires. Les réactions inflammatoires sont déclenchées lorsque les cellules immunitaires du système immunitaire inné sont activées, que ce soit par des agents pathogènes envahissants ou des lésions tissulaires. La libération de cytokines pro-inflammatoires par ces cellules active alors les membres du système immunitaire adaptatif pour déclencher une réponse inflammatoire.
L’administration de kétamine pendant ou avant les opérations chirurgicales a été utilisée pour obtenir un résultat postopératoire plus favorable, principalement en raison de ses actions visant à réduire la production de cytokines pro-inflammatoires en excès. Les effets anti-inflammatoires (c’est-à-dire la réduction des cytokines pro-inflammatoires) de doses subanesthésiques préopératoires de 0,15-0,25 mg/kg (bolus i.v. unique) de kétamine ont été décrits chez l’homme. Il a été démontré que la kétamine inhibait la production de cytokines pro-inflammatoires induite par une réaction immunitaire, notamment le facteur nucléaire κB, et diminuait les taux sanguins de facteur de nécrose tumorale-α, d’interleukine 6 (IL-6), de protéine C-réactive et/ou d’oxyde nitrique synthase inductible. La capacité de la kétamine à réduire les taux de cytokines pro-inflammatoires pourrait avoir une importance clinique, étant donné que des taux élevés d’IL-6 ont été associés à de mauvais résultats postopératoires. Toutefois, cette possibilité n’a pas encore fait l’objet d’une étude systématique.
Outre ses effets sur les cytokines pro-inflammatoires, la kétamine réduit la production d’oxyde nitrique induite par l’inflammation de manière dépendante de la dose. Les effets anti-inflammatoires de la kétamine ont été observés lorsque le médicament était administré avant et après une stimulation immunitaire, ce qui indique que la kétamine peut être capable de prévenir l’exacerbation de l’inflammation et de réduire l’inflammation existante. Il existe des preuves que la kétamine peut atténuer l’hyperalgésie postopératoire induite par un traumatisme en modulant la réponse inflammatoire, ce qui est bénéfique dans le contexte de la douleur postopératoire chronique.
Il a également été démontré que la kétamine corrigeait les marqueurs osseux inflammatoires anormaux dans les troubles dépressifs majeurs. En particulier, une perfusion i.v. de 40 minutes de kétamine (0,5 mg/kg) a augmenté les taux d’ostéoprotégérine – récepteur activateur du facteur nucléaire κB ligand – et d’ostéopontine – marqueurs prédictifs de l’inflammation osseuse – chez des patients souffrant d’un trouble dépressif majeur, mais n’a eu aucun effet chez des témoins sains. En outre, les taux sériques des cytokines pro-inflammatoires que sont le facteur de nécrose tumorale-α, l’interféron γ et l’interleukine 2, 5 et 10 n’ont pas été modifiés après une perfusion i.v. subanesthésique de 40 minutes de kétamine (0,5 mg/kg) chez des patients souffrant de dépression, tandis que les taux de la cytokine anti-inflammatoire IL-6 ont augmenté 230 minutes après la perfusion de kétamine. Cependant, cet effet de la kétamine sur les niveaux d’IL-6 n’était pas associé aux actions antidépressives du médicament. Il est possible que la perfusion elle-même ait entraîné une augmentation des niveaux d’IL-6 liée au stress aigu, étant donné que ce phénomène a également été observé après une perfusion de solution saline. Dans l’ensemble, ces résultats indiquent que les actions anti-inflammatoires de la kétamine se produisent principalement en présence d’une immunostimulation, alors que le médicament n’exerce aucun effet sur l’équilibre des cytokines en l’absence de réaction inflammatoire. Ainsi, la kétamine pourrait agir comme un immunomodulateur et non comme un agent immunosuppresseur, ce qui est particulièrement important car la kétamine est couramment administrée lors de l’induction de l’anesthésie, avant une intervention chirurgicale.
Les doses et les concentrations plasmatiques de kétamine utilisées pour obtenir des effets thérapeutiques cliniques sont répertoriées dans le tableau 1.
[TABLEAU 1]
B. Effets secondaires
1. Effets psychoactifs
a. Effets dissociatifs et psychotomimétiques
La kétamine exerce, en fonction de la dose, de vastes influences sur la conscience et la perception, certains patients faisant état de sensations dissociatives et extracorporelles (expériences/illusions hors du corps) lorsqu’ils se remettent d’une anesthésie induite par la kétamine. Si ces effets de la kétamine ont permis d’établir qu’il s’agissait d’un anesthésique dissociatif, les mêmes effets ont également été observés à des doses subanesthésiques.
Les effets psychoactifs les plus courants signalés après une seule administration i.v. subanesthésique de kétamine comprennent la dissociation (distorsions des stimuli visuels, auditifs ou somatosensoriels, ou altérations de la perception de soi ou du temps), des effets psychotomimétiques positifs (désorganisation conceptuelle, hallucinations, méfiance, contenu inhabituel des pensées) et des effets psychotomimétiques négatifs (affect émoussé, retrait émotionnel, ralentissement moteur). Ces effets ont été signalés à la fois dans des études contrôlées randomisées et des études non randomisées ou ouvertes. Par exemple, une étude randomisée, en double aveugle et contrôlée par placebo menée par Krystal et al. (1994) a montré qu’une perfusion i.v. de 40 minutes de la dose subanesthésique de 0,5 mg/kg de kétamine (Cmax résultante estimée à ∼100-250 ng/ml ou 0,42-1,1 µM) entraîne des aberrations perceptives qui correspondent à des états dissociatifs, ainsi que des symptômes psychotomimétiques positifs et négatifs. Ces effets sont apparus dans les 10 minutes suivant le début de la perfusion de kétamine et se sont atténués dans les 40 minutes suivant la fin du traitement. En revanche, peu ou pas d’effets psychoactifs ont été observés à la dose de 0,1 mg/kg (entraînant une concentration plasmatique de kétamine de ∼25-50 ng/ml ou 0,1-0,2 µM ; Krystal et al., 1994). Il a également été démontré que la kétamine (bolus de 0,3 mg/kg ; Cmax = ∼120 ng/ml ou 0,5 µM) exacerbe les symptômes psychotiques chez les patients souffrant de schizophrénie (Lahti et al., 2001). De même, Malhotra et al. (1997) ont également rapporté que la kétamine augmentait les symptômes psychotiques chez les patients souffrant de schizophrénie lorsqu’elle était administrée en bolus i.v. unique de 0,12 mg/kg, suivi d’une perfusion de 60 minutes de 0,65 mg/kg (dose totale de 0,77 mg/kg).
Les illusions et les altérations de l’audition, de la vision et de la proprioception ont été attribuées à l’action de la (S)-cétamine, tandis que les sensations de relaxation ont été associées à l’action de la (R)-cétamine. En particulier, à des doses équimolaires produisant des taux plasmatiques moyens de kétamine de 379 ± 71 ng/mg (soit 1,59 ± 0,30 μM) et de 389 ± 74 ng/mg (soit, 1,64 ± 0,31 μM) pour la (S)- et la (R)-cétamine, respectivement, l’énantiomère (S)-cétamine a provoqué des réactions psychotiques aiguës à un taux plasmatique moyen de kétamine de 539 ng/ml (soit 2,27 μM), alors que la (R)-cétamine n’a pas été associée à ces actions psychotomimétiques. En revanche, l’administration de (R)-cétamine a induit un sentiment de “bien-être” et un effet bénéfique sur l’humeur, mesuré par l’échelle d’évaluation de l’humeur Eigenschaftsworterliste (EWL) (Vollenweider et al., 1997). Une étude clinique menée par Mathisen et al. (1995) a montré que 56% des patients souffrant de douleurs orofaciales et traités avec de la (S)-cétamine (0,45 mg/kg, i.m. ; Cmax sérique = ∼120 ng/ml ou 0. 5 µM) ont eu des illusions, alors que seulement 22 % de ceux qui ont été traités avec de la (R)-cétamine ont eu des illusions, même si une dose plus élevée de (R)-cétamine a été utilisée (1,8 mg/kg, i.m. ; Cmax sérique = ∼590 ng/ml ou 2,5 µM). Dans cette étude, la prévalence des illusions chez les patients traités par la (S)-cétamine était comparable à celle observée chez les patients traités par la (R,S)-cétamine à une dose de 0,9 mg/kg, i.m. ; Cmax sérique = ∼297 ng/ml ou 1,25 µM. Des altérations de l’audition ont été signalées chez 78 %, 67 % et 57 % des patients traités par la (S)-, la (R)- et la (R,S)-cétamine, respectivement, tandis que des troubles de la vision ont été signalés par 100 %, 78 % et 85 % des patients recevant la (S)-, la (R)- et la (R,S)-cétamine, respectivement. En outre, le traitement par la (S)-cétamine a entraîné des troubles de la proprioception chez 100 % des patients, contre 56 % et 71 % des patients recevant respectivement de la (R)- ou de la (R,S)-cétamine. Bien que 43 % des patients traités par la (R,S)-cétamine aient fait état de rêves et d’hallucinations, aucun de ces effets n’a été signalé par les patients traités par la (S)- ou la (R)-cétamine.
Une étude menée chez des volontaires sains n’a révélé aucune différence entre les effets post-anesthésiques de la (S)- (140 ± 21 mg), de la (R)- (429 ± 37 mg) ou de la (R,S)-cétamine (275 ± 25 mg) quant à leur propension à provoquer des sensations de flottement (moyenne de 67 % des individus), de diplopie (vision double ; 60 %) ou d’étourdissement (47 % ; White et al., 1985). Ces effets se sont produits à des concentrations plasmatiques plus élevées de (R)-cétamine que de (S)- et (R,S)-cétamine.
b. Troubles de la mémoire et de la cognition
Outre les symptômes dissociatifs et psychotomimétiques, plusieurs études ont identifié des effets défavorables de l’administration subanesthésique de kétamine sur la cognition (voir également Ke et al., 2018). Des études ont rapporté que la kétamine diminue l’acuité mentale, la concentration, le rappel et la reconnaissance, ainsi que les formes explicites (épisodique et sémantique) et implicites (procédurale) de mémoire pendant ou peu après l’administration (pour les détails de la posologie, voir le tableau 1).
La vigilance, la fluidité verbale et le rappel différé sont également altérés pendant/immédiatement après une perfusion i.v. de 40 minutes de 0,5 mg/kg de kétamine (entraînant une Cmax plasmatique estimée à ∼100-250 ng/ml ou 0,42-1,1 µM) ; ces effets disparaissent peu après la fin de la perfusion. La fonction cognitive globale et le rappel immédiat semblent rester intacts pendant la perfusion de kétamine. D’après les résultats d’études transversales, l’abus de kétamine à long terme est également associé à des troubles cognitifs. Toutefois, en raison de la nature de ces études, il est difficile de contrôler pleinement l’impact d’autres facteurs comorbides ou environnementaux.
c. Abus
Alors que les effets psychotropes aigus de la kétamine peuvent provoquer un malaise chez certaines personnes, ses propriétés dissociatives l’ont rendue souhaitable pour un usage récréatif. Cependant, certains utilisateurs peuvent ressentir une agitation accrue ou des crises d’anxiété ou de panique. Dans les 10 minutes suivant le début d’une perfusion i.v. de 40 minutes d’une dose subanesthésique de 0,5 mg/kg de kétamine (entraînant une Cmax plasmatique estimée à ∼100-250 ng/ml ou 0,42-1,1 µM), des sujets sains ont déclaré se sentir “défoncés” (c’est-à-dire subjectivement comparables à ceux de l’intoxication alcoolique). Une dose plus faible de kétamine de 0,1 mg/kg (entraînant une Cmax plasmatique = ∼25-50 ng/ml ou 0,1-0,2 µM) a provoqué une légère euphorie (c’est-à-dire un bourdonnement).
Bien qu’il n’existe pas d’études contrôlées sur le potentiel d’abus de la kétamine, des informations précieuses sur les effets aigus et chroniques de la kétamine ont été tirées de rapports sur l’usage récréatif. En général, les doses utilisées pour la prise récréative de kétamine peuvent varier entre 1 et 2 mg/kg (i.v.), 50 et 150 mg (i.m.), 100 et 500 mg (oral), ou 30 et 400 mg (insufflation intranasale). Bien qu’il soit impossible de déterminer directement les effets des doses spécifiques utilisées à des fins récréatives en raison du manque d’études contrôlées les évaluant, les utilisateurs rapportent que des doses plus faibles induisent de légers effets stimulants, dissociatifs et hallucinogènes, tandis que des doses plus élevées provoquent des symptômes psychotomimétiques et une séparation de la réalité.
La voie d’administration récréative la plus courante est l’insufflation nasale, avec un début de sensation de “high” variant entre 5 et 10 minutes, et durant entre 40 et 75 minutes. Lorsque la consommation est maximale, les usagers rapportent que la kétamine induit une expérience hautement dissociative marquée par un état de conscience altéré et un détachement sensoriel (familièrement appelé le “k-hole”), que certains décrivent comme étant comparable à une expérience de mort imminente.
À des concentrations plasmatiques comprises entre 50 et 200 ng/ml (0,21-0,84 µM), la kétamine améliore de manière dose-dépendante la perception sensorielle (c.-à-d. l’intensité du son), la connexion émotionnelle et la capacité à s’adapter à l’environnement, intensité du son), la connexion émotionnelle, le sentiment d’irréalité et les expériences extracorporelles, et peut être associée à des hallucinations visuelles, à une altération de la perception de soi et du temps, et à des sensations de flottement. Les effets indésirables signalés par les consommateurs d’alcool illicite sont notamment des vertiges, une vision floue, des troubles de l’élocution, des vomissements, des palpitations et des douleurs thoraciques ; voir la section sur les effets périphériques ci-dessous. On a émis l’hypothèse que la diminution des sensations tactiles et musculo-squelettiques provoquée par la kétamine entraînait un sentiment d’apesanteur ou de détachement de soi, ce qui pourrait contribuer aux sensations extracorporelles. En outre, l’utilisation à long terme de la kétamine peut entraîner des flashbacks, des dysfonctionnements attentionnels et d’autres dysfonctionnements cognitifs, ainsi qu’une diminution de la sociabilité, mais la poursuite de l’utilisation est renforcée par les autres effets psychotropes. Malgré ses propriétés renforçantes, les cas de dépendance à la kétamine sont relativement rares, mais ont été signalés. Des données suggèrent également que l’utilisation répétée de la kétamine peut entraîner une tolérance à la drogue.
2. Effets périphériques directs et indirects
À des doses subanesthésiques (∼0,5 mg/kg administrés par voie i.v. en 40 minutes), la kétamine peut entraîner des perturbations vestibulaires, notamment des vertiges et des nausées/vomissements. L’action de la kétamine sur le système nerveux sympathique est associée à des effets cardiovasculaires généraux (p. ex. tachycardie, hypertension, palpitations) qui se manifestent tant en milieu clinique (0,5-1,0 mg/kg i.v.) qu’en milieu récréatif (100-200 mg i.m. ou s.c. ; Weiner et al., 2000). Bien que généralement considérée comme cliniquement insignifiante, une légère dépression respiratoire est rapportée à des doses allant de 0,39 à 3,0 mg/kg. En outre, les effets hémodynamiques (c’est-à-dire la pression artérielle et la fréquence cardiaque) n’ont pas varié de manière significative entre la (S)-, la (R)- et la (R,S)-cétamine (White et al., 1985), bien qu’au moins une étude suggère que la (S)-cétamine contribue spécifiquement aux effets cardiovasculaires de la (R,S)-cétamine, tels que l’augmentation de la pression artérielle. Dans l’ensemble, une analyse rétrospective récente portant sur des personnes ayant reçu 684 perfusions i.v. de kétamine (0,5 mg/kg en 40 minutes) indique que les modifications de la pression artérielle sont modestes, bien tolérées et cliniquement insignifiantes.
Des effets oculaires (p. ex. nystagmus, diplopie, dilatation) sont signalés dans des contextes récréatifs, ainsi qu’en clinique, à des doses subanesthésiques de kétamine (p. ex. 0,25 mg/kg i.v.). Certains effets oculaires (vision trouble) ont été principalement associés à la (S)-cétamine. En outre, des effets musculo-squelettiques (p. ex. myoclonie, contractions, spasmes, ataxie, fasciculation) ont été observés dans des cas d’abus de kétamine.
L’usage récréatif prolongé de la kétamine est associé à des complications urologiques telles que la dysurie, l’augmentation de la fréquence et de l’urgence des mictions, l’incontinence, la douleur, l’hématurie et la cystite ulcéreuse. Il a été suggéré que la kétamine pouvait avoir un impact négatif direct sur les cellules interstitielles de la vessie, la cystoscopie ayant révélé un érythème, un œdème et une inflammation épithéliale chez les utilisateurs de kétamine à long terme. En outre, la tomographie par ordinateur a révélé un épaississement marqué de la paroi de la vessie, un rehaussement de la muqueuse et une inflammation périvésicale associés à l’usage récréatif de la kétamine. Il existe au moins un rapport de cas où la kétamine subanesthésique (0,1 mg/kg par heure administrée par voie i.v. pendant 12 heures) a été associée à une urgence et une incontinence urinaires.
3. Effets à long terme
Étant donné que le maintien de l’efficacité thérapeutique de la kétamine nécessite souvent l’administration répétée du médicament, il est important d’examiner les effets secondaires qui peuvent être uniquement associés à l’exposition chronique à la kétamine. Les effets résultant d’un traitement à long terme à la kétamine sont soit mal définis, soit peu rapportés. À ce jour, l’abus répété de kétamine a été le plus régulièrement associé à des déficits durables liés à la mémoire. Les décès causés par une surdose de kétamine, en l’absence d’intoxication multidrogue, sont très rares, bien que des décès accidentels causés par des chutes de hauteur, une hypothermie extrême ou des accidents de voiture impliquant des personnes consommant de la kétamine aient été signalés.
Dans l’ensemble, il n’existe pas, à notre connaissance, de rapport faisant état d’une dose létale de kétamine chez l’homme. Néanmoins, chez le rat, l’administration intraveineuse de (R,S)-cétamine et de (S)-cétamine à la dose de 40 mg/kg a entraîné une létalité significative, alors que tous les animaux ayant reçu de la (R)-cétamine à la même dose ont survécu. Bien que ces résultats indiquent qu’il faut être prudent lors de l’utilisation à long terme de la kétamine, il existe des preuves que l’administration répétée de doses subanesthésiques de kétamine peut avoir des effets bénéfiques à long terme. Par exemple, il a été démontré que l’administration répétée de kétamine à des doses subanesthésiques améliorait les résultats cliniques dans les cas de dépression résistante au traitement. L’administration répétée de kétamine a également été associée à une atténuation de la dissociation, de la déréalisation et des vertiges induits par la kétamine au fil du temps. Néanmoins, des effets dissociatifs et psychotomimétiques ont été observés dans des études contrôlées randomisées examinant les effets d’une exposition répétée à la kétamine par voie i.v. subanesthésique.
La plupart des effets secondaires de la kétamine, sinon tous, dépendent de la dose, sont transitoires et se résorbent d’eux-mêmes. Toutefois, pour mieux évaluer l’utilité thérapeutique de la kétamine dans tous les contextes cliniques, les futures études devraient viser à évaluer systématiquement la sécurité et l’efficacité d’un traitement aigu ou chronique à la kétamine, en termes de résultats à court et à long terme.
4. Neurotoxicité
Avec l’apparition de nouvelles indications nécessitant l’administration répétée de kétamine (par exemple, les effets antidépresseurs), on craint des effets indésirables plus profonds du traitement, notamment l’induction de lésions d’Olney. Signalées pour la première fois en 1989, les lésions d’Olney se caractérisent par l’apparition de vacuoles dans le compartiment cytoplasmique de certaines populations neuronales, où la lyse des mitochondries a été signalée. Ces phénomènes de vacuolisation neuronale se produisent principalement dans le cortex cingulaire postérieur et le cortex rétrosplénial après l’administration d’antagonistes des récepteurs N-méthyl-D-aspartate (NMDAR) (PCP, MK-801 et kétamine, par exemple) à des rats. À faibles doses, la vacuolisation semble disparaître dans les 24 heures suivant l’administration, ce qui suggère que des lésions cellulaires permanentes ne se produisent pas lorsque des antagonistes non compétitifs (PCP, MK-801, kétamine et dextrorphane) ou compétitifs (CPP, CGS 19755 et CGP 37849) des NMDAR sont utilisés à des doses cliniquement pertinentes. Cependant, il reste possible que des doses élevées (ou peut-être l’administration répétée de faibles doses) d’antagonistes des NMDAR, tels que la kétamine, conduisent à des dommages sélectifs irréversibles. Par exemple, des études précliniques chez le rat ont montré que l’administration d’une dose élevée de MK-801 (c’est-à-dire 5 mg/kg) entraîne une nécrose dans un petit sous-ensemble de neurones – un effet qui était associé à une augmentation du taux de mortalité en fonction de l’âge. En outre, des études menées sur des primates non humains ont montré que l’administration quotidienne répétée de kétamine (1 mg/kg par jour, i.v.) : 1) réduit l’intégrité de la matière blanche dans les connexions fronto-thalamo-temporales, évaluée par imagerie du tenseur de diffusion après un traitement de 3 mois (Li et al., 2017), et 2) augmente la mort cellulaire dans le cortex préfrontal, évaluée par coloration de la digoxigénine-désoxyuridine nick-end marking médiée par la désoxynucléotidyl transférase terminale sur des coupes de cerveau obtenues chez des animaux traités pendant 6 mois.
On a constaté que la vacuolisation se produisait après l’administration de doses élevées (c’est-à-dire 40-60 mg/kg, s.c.) mais pas de doses faibles à modérées (5-20 mg/kg, s.c.) de kétamine chez les rats. Notamment, ces doses sont beaucoup plus élevées que les doses requises pour les actions analgésiques, anti-inflammatoires ou antidépressives du médicament. Par conséquent, la pertinence des lésions d’Olney pour l’utilisation répétée de kétamine chez l’homme est controversée et difficile à évaluer. Une étude d’imagerie par résonance magnétique a rapporté que les consommateurs récréatifs de kétamine (durée totale d’utilisation de la kétamine : 0,5-12 ans) présentaient une atrophie corticale dans les lobes frontal, pariétal et occipital, et que les atrophies mesurables étaient associées à un début d’utilisation de la drogue survenu 2-4 ans auparavant. En outre, une autre étude menée auprès d’usagers récréatifs (durée totale d’utilisation de la kétamine : 1-10,5 ans) a fait état d’une perte d’intégrité de la microstructure de la substance blanche corticale frontale qui était corrélée à la durée totale d’utilisation de la kétamine au cours de la vie.
Yeung et al. (2010) ont signalé la présence de cellules tau (protéine associée aux microtubules) hyperphosphorylées dans les cortex préfrontal et entorhinal de primates non humains et de souris ayant reçu des administrations quotidiennes de kétamine (1 mg/kg, bolus i.v. pour les singes et 30 mg/kg, injections i.p. pour les souris) sur une période de 3 à 6 mois. L’hyperphosphorylation de la protéine Tau a été associée au déclin de la mémoire observé chez les patients atteints de la maladie d’Alzheimer, ce qui pourrait indiquer un mécanisme sous-jacent aux troubles de la mémoire consécutifs à l’utilisation de la kétamine (voir la section Mémoire et troubles cognitifs). En outre, l’administration chronique intermittente de (S)-kétamine a entraîné une perte d’immunoréactivité de la parvalbumine dans l’hippocampe et le cortex préfrontal des souris, ce qui concorde avec les résultats obtenus dans des modèles animaux de psychose et de schizophrénie. Conformément à la plus faible puissance de la (R)-cétamine pour inhiber les NMDAR par rapport à l’énantiomère (S)-cétamine (voir la section Récepteurs du N-Méthyl-D-Aspartate), l’administration chronique intermittente de (R)-cétamine, contrairement à celle de la (S)-cétamine (toutes deux administrées à 10 mg/kg i.p., une fois par semaine pendant une période totale de 8 semaines chez la souris), n’a entraîné aucune perte de l’immunoréactivité de la parvalbumine. Dans l’ensemble, compte tenu de l’expansion des applications de la kétamine, il sera essentiel de mieux définir les effets à long terme de l’utilisation chronique de la kétamine.
Les doses et les concentrations plasmatiques de kétamine qui entraînent des effets secondaires indésirables chez l’homme sont répertoriées dans le tableau 1.
II. Pharmacocinétique
A. Métabolisme
La kétamine subit un métabolisme important (Fig. 1), d’abord par déméthylation de l’azote en norkétamine, une réaction catalysée principalement par les enzymes hépatiques CYP2B6 et CYP3A4 du cytochrome P450. La déméthylation de la kétamine se produit de manière stéréosélective, CYP3A4 déméthylant l’énantiomère (S)-cétamine plus rapidement que l’énantiomère (R)-cétamine, tandis que CYP2B6 déméthyle les deux énantiomères de la kétamine avec une efficacité presque égale (Portmann et al., 2010). La variabilité individuelle du métabolisme de la kétamine a été attribuée, en partie, aux différences d’expression des enzymes P450.
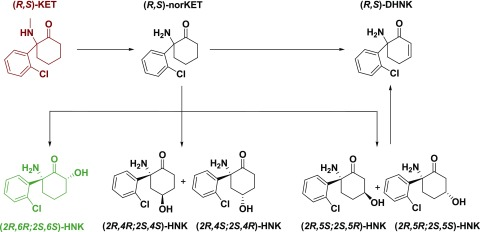
Après déméthylation de la kétamine en norkétamine, cette dernière est métabolisée en hydroxynorkétamines (HNK) et en déshydronorkétamine (DHNK) (Fig. 1). Les premières études ont montré que les HNK se forment par hydroxylation du cycle cyclohexyle de la norkétamine en divers endroits. Plusieurs de ces métabolites HNK ont été détectés chez l’homme après une perfusion de kétamine, les (2R,6R;2S,6S)-HNK et (2S,6R;2R,6S)-HNK étant les HNK circulants prédominants dans le plasma (Moaddel et al., 2010 ; Zarate et al., 2012a). Le métabolisme en (2R,6R;2S,6S)-HNK est principalement effectué par CYP2A6 et CYP2B6. Ces enzymes sont également responsables de la formation des (2S,4S;2R,4R)- et (2S,5S;2R,5R)-HNK. CYP3A4 et CYP3A5 sont les principales enzymes identifiées pour catalyser la conversion de la norkétamine en (2S,4R;2R,4S)-HNK, tandis que CYP2B6 est principalement responsable de la catalyse de la conversion de la norkétamine en (2S,5R;2R,5S)-HNK. L’autre métabolite secondaire est le DHNK. Le DHNK est directement formé à partir de la norkétamine, principalement sous l’action de l’enzyme CYP2B6, ou à partir du 5-HNK par déshydratation non enzymatique.
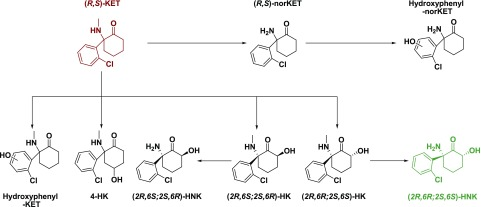
Outre les principales voies métaboliques de la kétamine, plusieurs autres voies ont également été étudiées (Fig. 2). L’une de ces voies est l’hydroxylation directe de la kétamine en 6-hydroxykétamine (HK). Le métabolisme de la kétamine en (2R,6R;2S,6S)-HK est principalement catalysé par CYP2A6, tandis que la production de (2S,6R;2R,6S)-HK est catalysée par les enzymes CYP3A4 et CYP3A5. La formation de (2R,6R;2S,6S)-HK est associée à une hydroxylation plus importante de la (S)-cétamine par rapport à la (R)-cétamine, ce qui suggère que cette réaction est énantiosélective. Le métabolite (2R,6R;2S,6S)-HK est facilement déméthylé par le CYP2B6 en HNK correspondant. Cependant, la déméthylation analogue du métabolite (2S,6R;2R,6S)-HK est signalée comme se produisant très lentement, avec une modeste contribution du CYP3A5. Outre les 6-HK, des preuves de la production du métabolite 4-HK ont été rapportées. Alors que l’hydroxylation de l’anneau phényle était initialement exclue du métabolisme de la kétamine, des études plus récentes ont fourni des preuves de la formation de ces métabolites hydroxyphénylés de la kétamine via l’action des enzymes CYP2C9 [principalement pour l’énantiomère (R)-cétamine] et de la mono-oxygénase contenant de la flavine [principalement pour l’énantiomère (S)-cétamine]. Enfin, des isomères phénoliques des HNK ont également été observés, potentiellement issus de l’hydroxylation de la norkétamine.
Un modèle pharmacocinétique de population a été construit pour la kétamine et ses métabolites chez des patients souffrant de dépression bipolaire résistante au traitement dans une étude qui a identifié la norkétamine, le DHNK et le (2R,6R;2S,6S)-HNK comme les principaux métabolites circulants dans le plasma après une perfusion i.v. unique de 40 minutes de kétamine (0,5 mg/kg). Il s’agit également des principaux métabolites identifiés dans le plasma de patients souffrant de dépression unipolaire ou bipolaire ou de CRPS et traités à la kétamine. Plus précisément, la norkétamine, le DHNK et le (2R,6R;2S,6S)-HNK ont été détectés dans le plasma de patients souffrant de dépression unipolaire ou bipolaire résistante au traitement dès 40 minutes après la fin de l’administration i.v. de kétamine (0,5 mg/kg administré en une seule perfusion de 40 minute). Le temps moyen nécessaire aux métabolites pour atteindre la concentration plasmatique maximale a été estimé à environ 1,33 heure pour la (R)- et la (S)-norkétamine et à 3,83 heures pour la (R)-DHNK, la (S)-DHNK et la (2R,6R;2S,6S)-HNK. Dans les échantillons de plasma de ces patients, les rapports entre la (S)- et la (R)-cétamine, la (S)- et la (R)-norkétamine, et la (S)- et la (R)-DHNK étaient respectivement de 0,84, 1,0 et 0,67, pendant une période de 40 à 230 minutes après la perfusion. À l’instar de ces résultats, l’administration i.v. de kétamine (2 mg/kg) à des patients ayant subi une intervention chirurgicale a entraîné un rapport plasmatique entre la (S)- et la (R)-kétamine de 0,91. En outre, des patients atteints de SRC recevant une perfusion i.v. continue de kétamine à une dose de 40 mg/h sur une période totale de 5 jours présentaient des rapports plasmatiques (S)-/(R)-cétamine, (S)-/(R)-norkétamine et (S)-/(R)-DHNK de 0,77, 0,71 et 0,71, respectivement.
Après une perfusion i.v. de kétamine de 40 minutes à la dose de 0,5 mg/kg chez des patients ayant reçu un diagnostic de trouble dépressif majeur résistant au traitement, les concentrations plasmatiques maximales étaient de 204,13 ± 101,46 ng/ml ou 0,86 ± 0,43 μM pour la kétamine (à 40 minutes), 73. 54 ± 31,86 ng/ml ou 0,33 ± 0,14 μM pour la norkétamine (à 80 minutes), 13,27 ± 6,92 ng/ml ou 0,06 ± 0,03 μM pour la DHNK (à 110 minutes), et 23,19 ± 11,88 ng/ml ou 0,097 ± 0,05 μM pour la (2R,6R;2S,6S)-HNK (à 230 minutes) ; voir le tableau 2. Chez les patients atteints de dépression bipolaire résistante au traitement, les concentrations plasmatiques maximales étaient de 177,23 ± 53,8 ng/ml ou 0,75 ± 0,23 μM pour la kétamine (à 40 minutes), 69,96 ± 19,98 ng/ml ou 0. 31 ± 0,09 μM pour la norkétamine (à 80 minutes), 50,5 ± 27,44 ng/ml ou 0,23 ± 0,12 μM pour la DHNK (à 110 minutes), et 37,59 ± 14,23 ou 0,16 ± 0,06 μM pour la (2R,6R;2S,6S)-HNK (tableau 2). Chez un patient atteint de CRPS recevant un traitement chronique à la kétamine (perfusion débutant à 10 mg/h, titrée à 40 mg/h, et durant 5 jours consécutifs), des taux plasmatiques significatifs de plusieurs métabolites HNK ont été détectés, les (2R,6R;2S,6S)- et (2R,6R;2S,6S)-HNK étant les principaux métabolites présents dans les échantillons obtenus au jour 3.
[TABLEAU 2]
Dans une étude menée par Cohen et al. (1973), les concentrations cérébrales des métabolites de la kétamine ont été mesurées après l’administration de kétamine (20 mg/kg) à des rats par la veine de la queue. Ces auteurs ont montré que la kétamine et la norkétamine s’accumulaient rapidement dans le cerveau, les concentrations maximales étant atteintes dans la minute suivant l’administration. Par la suite, il a été démontré que le (2R,6R;2S,6S)-HNK s’accumule également dans le tissu cérébral peu de temps après l’administration. L’injection intraveineuse dans la veine de la queue (perfusion de 2 minutes) de 20 mg/kg de (S)- ou (R)-cétamine à des rats a entraîné des niveaux cérébraux plus élevés de (2S,6S)-HNK par rapport à (2R,6R)-HNK, respectivement, avec des concentrations maximales de 769 ± 133 ng/g ou 3. 21 ± 0,55 μmol/kg à 20 minutes pour la (2S,6S)-HNK et 274 ± 47 ng/g ou 1,14 ± 0,20 μmol/kg à 10 minutes pour la (2R,6R)-HNK (Moaddel et al, 2015b ; tableau 2). L’hypothèse a été émise que cette différence était due à un processus d’absorption passive de ces métabolites dans le cerveau. Un rapport ∼1:1 pour les niveaux plasma/cerveau du (2R,6R;2S,6S)-HNK correspondant a été observé, indiquant que la pénétration de la barrière hémato-encéphalique ou le processus de transport dans le système nerveux central n’était pas médié par un processus énantiosélectif. Il est important de noter qu’aucun métabolisme in situ n’a été observé lorsque la kétamine a été incubée avec des microsomes de cerveau de rat (fraction S9). De même, les métabolites de la kétamine étaient inférieurs aux niveaux détectables dans le cerveau des souris après une administration intracérébroventriculaire in vivo de kétamine (P.Z., R.M., J.N.H., T.D.G., données non publiées), un résultat qui suggère que le métabolisme local de la kétamine ne se produit pas dans le cerveau.
Chez la souris, la norkétamine, le DHNK et les métabolites (2R,6R;2S,6S)-HNK ont été détectés dans le plasma dans les 10 minutes suivant l’administration i.p. de 10 mg/kg de kétamine. Les concentrations plasmatiques maximales étaient de 561,89 ± 86,09 ng/ml ou 2,36 ± 0,18 μM à 10 minutes pour la kétamine, 1098,89 ± 216,89 ng/ml ou 4,91 ± 0,97 μM à 10 minutes pour la norkétamine, 83. 92 ± 53,63 ng/ml ou 0,38 ± 0,24 μM à 30 minutes pour la DHNK, et 674,59 ± 278,23 ng/ml ou 2,81 ± 1,16 μM à 10 minutes pour la (2R,6R;2S,6S)-HNK, comme le résume le tableau 2. Dans le cerveau, la kétamine (1162,34 ± 202,05 ng/g ou 4,89 ± 0,85 μmol/kg de tissu), la norkétamine (450,94 ± 199,7 ng/g ou 2,02 ± 0,89 μmol/kg de tissu) et le (2R,6R;2S,6S)-HNK (498,35 ± 50,99 ng/g ou 2,08 ± 0,21 μmol/kg de tissu) ont été détectés dans les 10 minutes suivant l’administration de kétamine. La concentration maximale de kétamine dans le cerveau était supérieure de 51,66 % à la concentration plasmatique correspondante, tandis que les concentrations de norkétamine et de (2R,6R;2S,6S)-HNK dans les tissus cérébraux étaient respectivement inférieures de 58,96 % et de 26,13 % aux concentrations plasmatiques maximales correspondantes. Les niveaux de DHNK dans le tissu cérébral étaient inférieurs aux limites de quantification, ce qui concorde avec les observations selon lesquelles la DHNK se répartit dans les globules rouges et pénètre mal la barrière hémato-encéphalique.
B. Absorption
La kétamine est administrée à l’homme par de multiples voies, notamment i.v., i.m., orale, intranasale, épidurale et intrarectale. La voie d’administration la plus courante est la perfusion i.v., qui permet d’atteindre rapidement des concentrations plasmatiques maximales. L’administration intramusculaire, utilisée dans les cas d’urgence chez les patients non coopératifs, les nouveau-nés et les enfants, présente une biodisponibilité élevée de 93 %, les concentrations plasmatiques maximales étant atteintes dans les 5 à 30 minutes suivant l’administration ; toutefois, une analyse pharmacocinétique de population a fait état d’une biodisponibilité beaucoup plus faible après l’administration i.m. de kétamine chez les enfants (41 %). En revanche, la biodisponibilité orale de la kétamine est limitée à 16 %-29 %, les concentrations maximales du médicament étant atteintes en 20-120 minutes, en raison d’un métabolisme hépatique de premier passage important. La biodisponibilité orale de la (S)-cétamine a été calculée comme étant de 8 % à 11 %, ce qui correspond au métabolisme de premier passage plus important de la (S)-cétamine par rapport à la (R,S)-cétamine. La biodisponibilité de la kétamine par voie intranasale et intrarectale est respectivement de 45 % à 50 % et de 25 % à 30 %. L’administration intranasale est considérée comme une alternative intéressante à l’administration i.v. de kétamine car elle est moins invasive, entraîne une absorption systémique rapide et n’est pas soumise au métabolisme hépatique de premier passage.
Après administration orale de (2S,6S)-HNK à des rats (20 mg/kg), les concentrations plasmatiques maximales ont été atteintes en 0,4 ± 0,1 heure. La biodisponibilité orale de la (2S,6S)-HNK a été estimée à 46,3 % chez le rat. Chez la souris, la biodisponibilité orale du (2R,6R)-HNK est estimée à environ 50 % à la dose de 50 mg/kg (P.Z., R.M., J.N.H., T.D.G., données non publiées). La biodisponibilité orale des autres métabolites de la kétamine reste à déterminer.
C. Distribution
La kétamine est rapidement distribuée dans les tissus très perfusés, y compris le cerveau, et se lie aux protéines plasmatiques à hauteur de 10 à 50 %, ce qui se traduit par un important volume de distribution à l’état d’équilibre (Vd = 3-5 l/kg). Une seule administration i.v. en bolus d’une dose anesthésique de kétamine racémique chez l’homme (2 mg/kg) conduit à des concentrations plasmatiques égales de (S)-cétamine et de (R)-cétamine 1 minute après l’administration (Cmax = ∼1800 ng/ml ou 7,6 µM – estimation d’après Geisslinger et al., 1993). Cependant, l’administration i.v. (bolus) de 1 mg/kg de (S)-cétamine a entraîné une concentration plasmatique plus élevée du médicament 1 minute après la perfusion (Cmax = ∼2600 ng/ml : 11 µM – estimation d’après Geisslinger et al., 1993). Ces résultats sont particulièrement importants lorsqu’on compare les résultats de la (S)-cétamine à ceux de la kétamine racémique ou de la (R)-cétamine, car des doses plus faibles de (S)-cétamine sont nécessaires pour produire des concentrations de kétamine similaires ou supérieures dans le plasma. Notamment, il n’y a pas d’interconversion entre la (S)- et la (R)-cétamine, car l’administration de (S)-cétamine n’entraîne pas la formation de (R)-cétamine in vivo, et vice versa. Le plasma de patients souffrant de dépression bipolaire résistante au traitement, qui ont été traités par une perfusion i.v. de 40 minutes de 0,5 mg/kg de (R,S)-cétamine, présentait un rapport entre (S)- et (R)-cétamine de 0,84, avec des concentrations maximales de kétamine de 177,23 ± 53,8 ng/ml ou 0,75 ± 0,23 μM.
Chez la souris, l’administration de doses subanesthésiques de (S)- ou de (R)-cétamine (10 mg/kg, i.p.) a entraîné des concentrations cérébrales similaires des deux médicaments [aire sous la courbe (ASC)dernière = 483,1 heures.ng/ml ou 2,03 heures.μmol/kg contre 591,9 heures. ng/ml ou 2,48 heures.μmol/kg, respectivement], les concentrations maximales étant Cmax = 1743 ± 560,6 ng/g ou 7,33 ± 2,36 μmol/kg pour la (S)-cétamine et 1886 ± 459,6 ng/ml ou 7,93 ± 1,93 pour la (R)-cétamine 10 minutes après l’injection. De même, il n’y avait pas de différence entre les taux de (S)-cétamine (Cmax = 2732 ± 535 ng/ml ou 11,49 ± 2,25 μM) et de (R)-cétamine (Cmax = 3430 ± 400 ng/ml ou 14,4 ± 1,68 μM) dans le plasma des rats 10 minutes après une administration i.v. de 20 mg/kg de chacun de ces énantiomères.
L’administration i.p. directe de (2R,6R)-HNK et de (2S,6S)-HNK chez la souris entraîne un rapport 1:1 entre les concentrations circulantes dans le plasma et dans le tissu cérébral (tableau 2), avec des concentrations totales (AUClast) plus élevées de (2S,6S)-HNK observées dans le plasma et le tissu cérébral par rapport à celles de (2R,6R)-HNK (cerveau : 7. 55 contre 3,05 h.μmol/kg ; plasma : 11,60 contre 3,22 h.μM ; tableau 2). Après administration i.v. de (2S,6S)-HNK chez le rat (20 mg/kg), l’exposition totale au médicament a été calculée comme suit : AUClast = 28 981 ± 6162 h.ng/ml ou 120,91 ± 25,71 h.μM, avec un volume de distribution Vd = 7,35 ± 0,74 l/kg. Après administration orale de (2S,6S)-HNK à des rats (20 mg/kg), l’exposition totale au médicament était de AUClast = 10 120 ± 1313 h.ng/ml ou 42,22 ± 5,48 h.μM.
D. Élimination
Bien que les concentrations plasmatiques de kétamine soient inférieures aux limites détectables dans un délai d’un jour après l’administration par voie intraveineuse d’une dose antidépressive de kétamine (0,5 mg/kg en perfusion de 40 minutes), des concentrations circulantes de DHNK et de (2R,6R;2S,6S)-HNK ont été observées jusqu’à trois jours après la perfusion de kétamine chez des patients ayant reçu un diagnostic de dépression bipolaire ou de dépression majeure résistante au traitement. La norkétamine et la kétamine ont été détectables jusqu’à 14 et 11 jours, respectivement, dans l’urine d’enfants ayant reçu des doses anesthésiques de kétamine, avec des concentrations rapportées de 0,1-1442 ng/ml (ou 0,0004-0,031 μΜ) pour la norkétamine et de 2-1204 ng/ml (ou 0,008-5,06 μM) pour la kétamine.
Chez l’homme adulte, la kétamine présente un taux de clairance élevé et une demi-vie d’élimination courte (2-4 heures). White et al. (1985) ont également mis en évidence une demi-vie d’élimination courte (155-158 minutes) pour la (S)-cétamine et la (R)-cétamine. L’élimination de la kétamine se fait principalement par les reins, avec de faibles niveaux excrétés sous forme de kétamine (2 %), de norkétamine (2 %) et de DHNK (16 %). La majorité du médicament (∼80 %) est excrétée sous forme de conjugués de HK et HNK labiles à l’acide glucuronique, qui sont éliminés dans l’urine et la bile.
Chez l’homme adulte, la demi-vie plasmatique terminale et les taux de clairance de la kétamine ne diffèrent pas de manière significative entre les voies d’administration i.v. (demi-vie = 186 minutes ; clairance corporelle totale = 19,1 ml/min par kilogramme) et intramusculaire (demi-vie = 155 minutes ; clairance corporelle totale = 23,2 ml/min par kilogramme). Cependant, il existe des preuves que l’administration répétée de kétamine prolonge son temps d’élimination. Par exemple, Adamowicz et Kala (2005) ont rapporté que, dans trois cas de perfusions uniques de kétamine par voie i.v. sur une période de deux ans (doses comprises entre 0,75 et 1,59 mg/kg), l’élimination de la kétamine a été ralentie de deux jours après la première perfusion à cinq jours après la deuxième et à onze jours après la troisième. L’élimination de la norkétamine est restée constante (c’est-à-dire 5 jours après chaque perfusion).
Par rapport aux adultes, la kétamine est éliminée environ deux fois plus vite chez les enfants. Ceci est en accord avec les preuves soutenant une durée d’anesthésie plus longue chez les adultes que chez les enfants après l’administration i.m. de 6 mg/kg de kétamine. En outre, une corrélation négative entre l’âge et la dose de kétamine par poids corporel nécessaire à l’anesthésie a été rapportée chez les enfants. Ces différences pourraient être dues à des différences dans le métabolisme enzymatique de la kétamine chez les enfants par rapport aux adultes.
Chez l’homme, la (S)-cétamine a une demi-vie d’élimination légèrement plus longue que la kétamine racémique [∼5 heures pour la (S)-cétamine contre 2-4 heures pour la kétamine racémique], et sa clairance systémique est plus rapide lorsqu’elle est administrée seule que lorsqu’elle est administrée dans le mélange racémique [26,3 ± 3,5 ml/kg par minute pour la (S)-cétamine contre 14,8 ± 1,7 ml/kg par minute lorsqu’elle est administrée sous forme de kétamine racémique]. Cela peut suggérer une inhibition de la clairance de la (S)-cétamine par l’énantiomère de la (R)-cétamine lorsque le mélange racémique est administré. Cette inhibition pourrait contribuer à l’allongement du temps de réveil chez les patients recevant de la kétamine racémique par rapport à ceux recevant de la (S)-cétamine. Nous notons que la clairance systémique de la (R)-cétamine après l’administration du racémate de kétamine est de 13,8 ± 1,3 ml/kg par minute, ce qui est similaire à l’énantiomère (S)-cétamine (14,8 ± 1,7 ml/kg par minute).
Après administration i.v. de (2S,6S)-HNK à des rats (20 mg/kg), le taux de clairance a été calculé comme étant de 704 ± 139 ml/kg par heure, avec une demi-vie d’élimination de 8,0 ± 4,0 heures. L’administration orale de ce métabolite a entraîné une demi-vie d’élimination de 3,8 ± 0,6 heures.
Dans l’ensemble, il est important de noter qu’il existe d’importantes différences entre les espèces en ce qui concerne les valeurs de demi-vie, les ASC, les Cmax et les taux de clairance de la kétamine et de ses métabolites (voir le tableau 2). Il convient d’en tenir compte lors de la comparaison des effets comportementaux de schémas posologiques spécifiques de la kétamine et de ses métabolites chez la souris, le rat et l’homme. Néanmoins, les concentrations cérébrales de kétamine et de ses métabolites après l’administration de kétamine chez l’homme ne sont pas connues et, par conséquent, les comparaisons directes ne sont pas évidentes.
III. Pharmacodynamie de la kétamine et de ses métabolites
Comme indiqué plus haut, la kétamine est un antagoniste des NMDAR, et ses effets analgésiques et anesthésiques bien caractérisés sont principalement attribués à l’inhibition des NMDAR. Cependant, les cibles pharmacologiques de la kétamine ne se limitent pas aux NMDAR. Il a été rapporté que la kétamine interagit avec plusieurs autres récepteurs et canaux ioniques, notamment les récepteurs dopaminergiques, sérotoninergiques, sigma, opioïdes et cholinergiques, ainsi que les canaux activés par l’hyperpolarisation et gérés par les nucléotides cycliques (HCN). La kétamine a généralement une affinité plus faible (constante inhibitrice Ki-plus élevée) pour ces récepteurs et canaux que pour les NMDAR, et des laboratoires indépendants n’ont pas validé un grand nombre des résultats rapportés.
Les premières études pharmacodynamiques de la (R,S)-cétamine ont été menées chez le rat et ont porté sur les effets anesthésiques de la molécule mère et de ses deux principaux métabolites, la (R,S)-norkétamine et la (2R,6R;2S,6S)-HNK. Les résultats ont montré qu’une administration i.v. en bolus de 40 mg/kg de (R,S)-cétamine et de (R,S)-norkétamine produisait des effets anesthésiques et augmentait l’activité locomotrice spontanée pendant la phase de récupération postanesthésique, alors que le (2R,6R;2S,6S)-HNK (même dose) n’avait aucun effet anesthésique ou hyperlocomoteur. Par conséquent, la (2R,6R;2S,6S)-HNK a été décrite comme un métabolite inactif, et la majorité des évaluations pharmacodynamiques ont été réalisées uniquement avec la (R,S)-cétamine et la (R,S)-norkétamine. Cependant, il a été récemment démontré que les métabolites HNK de la kétamine sont biologiquement actifs. Il a été démontré que les métabolites (2S,6S)- et (2R,6R)-HNK exercent des réponses comportementales antidépressives chez les rongeurs. Conformément aux actions antidépressives plus puissantes de la (R)-cétamine par rapport à l’énantiomère (S)-cétamine, la (2R,6R)-HNK s’est avérée être un antidépresseur plus puissant que la (2S,6S)-HNK dans plusieurs tests sur les animaux.
A. Récepteurs N-Méthyl-D-Aspartate
Historiquement, le principal récepteur cible reconnu de la kétamine est le NMDAR, pour lequel la kétamine agit comme un bloqueur non compétitif de canal ouvert. Les NMDAR sont des canaux ioniques glutamatergiques constitués de différentes combinaisons de quatre sous-unités codées par l’un des sept gènes : GluN1, GluN2A-D et GluN3A-B. Les NMDAR sont très perméables aux ions calcium, qui peuvent déclencher l’activation d’un certain nombre de voies intracellulaires dans les neurones et les cellules gliales. Au repos, les canaux NMDAR sont bloqués de manière tonique par le magnésium (Mg2+). L’activation efficace des récepteurs nécessite les éléments suivants : 1) une dépolarisation de la membrane, qui déplace le blocage du Mg2+, et 2) la fixation du glutamate et du coactivateur glycine et/ou D-sérine.
La kétamine a été initialement caractérisée comme un antagoniste du NMDAR par David Lodge et ses collègues, une découverte qui a été confirmée par la suite par d’autres chercheurs. La kétamine se lie au site allostérique de la phencyclidine (PCP) situé dans le pore du canal du NMDAR et bloque ainsi le récepteur de manière non compétitive. La kétamine a une capacité de piégeage relativement élevée (∼86 %) (liaison dans le pore du canal ionique après la fermeture du canal) pour bloquer les NMDAR, en se liant au même site que le PCP (>98 % de piégeage) et le MK-801 (100 % de piégeage). L’affinité de liaison de la kétamine au site de liaison du PCP se situerait entre 0,18 et 3,1 μM en présence de Mg2+ (tableau 3).
[TABLEAU 3]
Le blocage des NMDAR serait à l’origine des effets anesthésiques dissociatifs et amnésiques de la kétamine, ainsi que des effets antidépresseurs, analgésiques et psychotomimétiques induits par la drogue. On suppose également que les déficits cognitifs induits par la kétamine sont dus à l’inhibition des NMDAR. La (S)-kétamine a une affinité/puissance pour le site PCP du NMDAR environ quatre fois supérieure à celle de l’isomère (R), et deux fois supérieure à celle du mélange racémique [(S)-kétamine : Ki = 0,3-0,69 μM ; (R)-cétamine : Ki = 1,4-2,57 μM ; et (R,S)-cétamine : Ki = 0,18-3,1 μM, en présence de Mg2+ extracellulaire]. Les effets de la (S)-cétamine et de la (R)-cétamine ont également été évalués sur les courants cationiques activés par les récepteurs NMDA des neurones hippocampiques de rat cultivés à cellules entières et clampés en tension. Ces auteurs ont montré que les deux énantiomères bloquaient les courants NMDAR en fonction du voltage et de l’utilisation, la (S)-cétamine étant environ deux fois plus puissante que la (R)-cétamine (IC50 = 0,80 contre 1,53 μM, respectivement). En outre, la (S)-cétamine a un pouvoir d’inhibition des courants provoqués par le NMDA dans les neurones de la corne dorsale du chat 2,5 à 3 fois supérieur à celui de l’énantiomère (R)-cétamine. Cette affinité/puissance supérieure de l’isomère (S)-cétamine expliquerait pourquoi la (S)-cétamine est un anesthésique plus puissant que la (R,S)-cétamine. Conformément à ces différences stéréospécifiques de puissance d’inhibition du NMDAR par les isomères de la kétamine, la valeur ED50 pour l’induction de l’hypnose (perte du réflexe de redressement) était plus faible pour la (S)-cétamine et la (R,S)-cétamine (3,5 et 5,6 mg/kg, respectivement) que pour la (R)-cétamine (10,3 mg/kg). De même, Ryder et al. (1978) ont montré que la (S)-cétamine est un analgésique ∼3 fois plus puissant, un hypnotique 1,5 fois plus puissant (perte du réflexe de redressement) et un stimulant locomoteur 1,8 fois plus puissant que la (R)-cétamine. En particulier, les doses analgésiques efficaces médianes (s.c.) se sont révélées être de 6,5, 3,7 et 11 mg/kg pour la (R,S)-cétamine, la (S)-cétamine et la (R)-cétamine, respectivement. Les doses hypnotiques médianes pour la (R,S)-cétamine, la (S)-cétamine et la (R)-cétamine ont été calculées comme étant respectivement de 45, 38 et 56 mg/kg. En outre, la (S)-cétamine (25 mg/kg, s.c.) a induit une perturbation plus profonde du déclenchement sensorimoteur que l’énantiomère (R)-cétamine (25 mg/kg, s.c.) dans le paradigme de l’inhibition de la préimpulsion chez le rat, bien que la (R)-cétamine ait également montré un effet subtil dans cette étude par rapport aux rats traités par le témoin. En accord avec ces résultats, Yang et al. (2015) ont montré que l’administration de (S)-cétamine, mais pas de (R)-cétamine chez la souris, entraînait une perturbation de la synchronisation sensorimotrice et de l’hyperlocomotion. Des concentrations subanesthésiques de kétamine (perfusion i.v. de 40 minutes ; 0,5 mg/kg), qui exercent des actions antidépressives chez des patients souffrant de dépression majeure, ont entraîné une occupation maximale de 31 % ± 18 % des NMDAR. Cette occupation est similaire à l’occupation NMDAR estimée (32 % ± 6 % maximum) à la suite d’une dose de kétamine antidépressive chez le rat (10 mg/kg, i.p.). Néanmoins, la (R)-cétamine s’est révélée être un antidépresseur plus puissant et plus durable que l’énantiomère (S)-cétamine dans plusieurs modèles de rongeurs, en utilisant une gamme de doses 30 fois plus élevée. Il ne semble pas y avoir de différences dans l’exposition cérébrale des deux énantiomères, ce qui remet en question l’hypothèse de l’inhibition des NMDAR comme seul médiateur des effets antidépresseurs de la kétamine.
Dans les fractions membranaires d’homogénats de cerveau humain post-mortem, les valeurs IC50 pour le déplacement du [3H]MK-801 par la (S)- et la (R)-cétamine ont été rapportées comme étant respectivement de 1,6-1,9 et de 7,2-10 μM, en présence de Mg2+ extracellulaire. De même, dans le tissu cortical de rat, la (S)-cétamine a inhibé les courants évoqués par le NMDA (10 μΜ) avec une CI50 de 0,9 ± 1,4 μM, alors que la (R)-cétamine était un inhibiteur moins puissant avec une CI50 de 3,0 ± 1,4 μM (Ebert et al., 1997). Des enregistrements électrophysiologiques en patch-clamp à cellules entières obtenus à partir de cellules rénales embryonnaires humaines (HEK)293T transfectées avec différentes sous-unités NMDAR ont révélé qu’en l’absence de Mg2+ extracellulaire, la kétamine inhibe les NMDARs contenant GluN1/GluN2A (IC50 = 0. 33 ± 0,01 μM) et GluN1/GluN2B (IC50 = 0,31 ± 0,02 μM) avec une puissance légèrement supérieure à celle des sous-unités GluN1/GluN2C (IC50 = 0,51 ± 0,01 μM) et GluN1/GluN2D (IC50 = 0,83 ± 0,02 μM) (Kotermanski et Johnson, 2009). En revanche, en présence de niveaux physiologiques de Mg2+ (1 mM), la kétamine bloque les NMDAR contenant les sous-unités GluN1/GluN2C (IC50 = 1,18 ± 0,0 μM) et GluN1/GluN2D (IC50 = 2,95 ± 0. 02 μM), avec une puissance supérieure à celle des sous-unités GluN1/GluN2A (IC50 = 5,35 ± 0,34 μM) et GluN1/GluN2B (IC50 = 5,08 ± 0,02 μM). Néanmoins, Yamakura et al. (1993) n’ont pas réussi à identifier de différences dans l’inhibition induite par la kétamine des différentes sous-unités des récepteurs NMDAR dans des ovocytes de Xénope injectés avec des ARNm spécifiques de sous-unités synthétisées in vitro. Ces résultats soulignent le manque de clarté quant aux effets différentiels de la kétamine sur les sous-types de NMDAR composés de différentes sous-unités.
Des études ont montré que la (S)-cétamine inhibe les NMDAR composés de GluN1/GluN2C (IC50 = 1,11 μM) et de GluN1/GluN2D (IC50 = 1,50 μM) avec une puissance plus élevée que ceux composés de GluN1/GluN2A (IC50 = 16,10 μM) en présence de 2 mM de Mg2+. La capacité de la (S)-cétamine à inhiber GluN1/GluN2B (CI50 = 1,55 μM) serait similaire à sa capacité à inhiber les NMDAR contenant GluN1/GluN2C et GluN1/GluN2D en présence de 2 mM de Mg2+. Ces résultats indiquent que toute puissance préférentielle de la kétamine n’est probablement pas le résultat d’une plus grande affinité de la kétamine à se lier aux GluN2C-NMDARs en soi, mais peut être due à une capacité différentielle de liaison au Mg2+, ou à des interactions entre le médicament et le Mg2+ dans le canal. Ainsi, la kétamine peut bloquer de manière différenciée des sous-types spécifiques de NMDAR dans le cerveau en fonction des concentrations locales de Mg2+. À l’appui de ce concept, en l’absence de Mg2+, la kétamine bloque les NMDAR contenant GluN2B avec une puissance supérieure à celle des NMDAR contenant d’autres sous-unités GluN2, comme cela a été mesuré à l’aide de sous-unités NMDAR GluN2A-D recombinantes exprimées dans des ovocytes de Xenopus.
En présence de Mg2+ extracellulaire, le métabolite N-déméthylé de la kétamine, la norkétamine, inhibe également le NMDAR. La (S)-norkétamine a un Ki rapporté de 1,70-2,25 μM pour les NMDAR dans la moelle épinière et le cortex cérébral, tandis que la (R)-norkétamine a une affinité de liaison environ huit fois plus faible (Ki = 13,0-26,46 μM ; Ebert et al., 1997 ; Moaddel et al., 2013) ; voir également le tableau 3. Conformément à ces résultats, la (S)-norkétamine (IC50 = 3,0 ± 0,8 μM) a inhibé plus puissamment les courants évoqués par les NMDA (10 μΜ)que la (R)-norkétamine (IC50 = 39 ± 1,4 μM) dans les neurones corticaux cérébraux de rat. Par conséquent, l’inhibition des NMDAR étant considérée comme le principal mécanisme d’action de la kétamine, les effets cliniques du médicament ont d’abord été attribués à la kétamine et à la norkétamine.
Les métabolites DHNK et HNK ont une capacité faible ou nulle à déplacer la liaison du [3H]MK-801 aux NMDAR. Le (R)-DHNK a une affinité plus faible que le (S)-DHNK (59,7-74,6 et 39,0-42,0 μM, respectivement) pour déplacer la fixation du [3H]MK-801 sur les NMDAR (Moaddel et al., 2013 ; Morris et al., 2017). Le (2S,6S)-HNK a un Ki = 10,4-21,0 μM pour déplacer la fixation du [3H]MK-801, tandis que le (2R,6R)-HNK ne se lie pas au site NMDAR-PCP avec une affinité appréciable (Ki > 100 μM). En outre, à des concentrations allant jusqu’à 10 μM, ni le (2S,6S)-HNK ni le (2R,6R)-HNK n’inhibent fonctionnellement les courants évoqués par les NMDAR dans les interneurones hippocampiques de rat. L’absence d’inhibition fonctionnelle des NMDAR par la (2R,6R)-HNK à 10 μM a également été rapportée par Suzuki et al. (2017). À une concentration plus élevée (50 μM), le (2R,6R)-HNK a modérément (∼40 %) inhibé les courants postsynaptiques excitateurs miniatures médiés par les NMDAR et enregistrés à partir de neurones hippocampiques en culture en l’absence de Mg2+. Ce résultat a étayé l’affirmation selon laquelle, à des concentrations supérieures à celles utilisées pour le traitement antidépresseur et en l’absence de Mg2+, le (2R,6R)-HNK pourrait inhiber fonctionnellement les NMDAR. Notamment, à la même concentration (50 μM) et dans les mêmes conditions expérimentales, la kétamine a induit une inhibition de >90 % des courants postsynaptiques excitateurs miniatures médiés par les NMDAR et enregistrés à partir de neurones hippocampiques (Suzuki et al., 2017). Les (2R,6S)-, (2S,6R)-, (2R,5R)-, (2S,5S)-, (2S,5S)-, (2R,5S)-, (2S,5R)-, (2R,4S)-, (2S,4R)-, (2R,4R)- et (2S,4S)-HNK n’ont pas d’affinité significative pour déplacer la fixation du [3H]MK-801 (Ki > 100 μM).
Il existe également des preuves qu’en réduisant les taux extracellulaires de D-sérine, les énantiomères de la kétamine et ses métabolites peuvent indirectement diminuer l’activation des NMDAR. La D-sérine, un coagoniste endogène des NMDAR qui se lie au site glycineB, est nécessaire à l’activation du complexe NMDAR et est produite par l’énantioconversion enzymatique de la L-sérine catalysée par la sérine racémase (Wolosker et al., 2008). L’incubation des cellules PC-12 avec des concentrations croissantes de (S)- et (R)-cétamine a exercé des effets différentiels sur les niveaux intracellulaires et extracellulaires de D-sérine. Plus précisément, l’application de (S)-cétamine a été associée à une augmentation de la D-sérine intracellulaire (CE50 = 0,82 ± 0,29 μM) et à une diminution des niveaux extracellulaires de D-sérine (CI50 = 0,82 ± 0,29 μM ; Singh et al., 2015). En revanche, la (R)-cétamine a diminué les concentrations intracellulaires (IC50 = 0,94 ± 0,16 μM) et extracellulaires de D-sérine (IC50 = 0,70 ± 0,10 μM ; Tableau 3). Des résultats similaires ont été observés en utilisant des cellules 1321N1 et des cellules neuronales primaires hippocampiques et corticales. Singh et al. (2015) ont également démontré que l’inhibition du transporteur d’acides aminés, ASCT2, entraînait des effets qualitativement similaires à ceux induits par la (S)-cétamine sur les taux de D-sérine. En outre, la coïncidence avec des concentrations sous-saturantes d’un inhibiteur de l’ASCT2 et de la (S)-cétamine a entraîné un effet additif dans les cellules PC-12 et les cellules neuronales primaires en ce qui concerne les niveaux de D-sérine, ce qui indique que les effets de la (S)-cétamine pourraient être dus à une inhibition des systèmes de transport d’acides aminés.
Les effets différentiels des énantiomères de la kétamine sur les taux de D-sérine pourraient contribuer à leurs effets comportementaux différentiels. En effet, alors que la (S)-cétamine est un médicament anesthésique et analgésique plus puissant que la (R)-cétamine, la (R)-cétamine est un antidépresseur plus puissant et plus durable que la (S)-cétamine dans plusieurs tests sur les animaux. En fait, la D-sérine joue un rôle dans la plasticité synaptique, et les taux plasmatiques de base de D-sérine sont négativement corrélés à la réponse au traitement par la kétamine chez les patients souffrant de dépression majeure, ce qui indique un rôle possible des taux de D-sérine dans les réponses antidépressives de la kétamine. In vivo, il a été démontré que l’administration subchronique (14 jours) de kétamine à des rats réduisait les niveaux d’ARNm de la sérine racémase dans le cerveau antérieur. Cependant, une administration unique de kétamine à la dose de 50 mg/kg a entraîné une augmentation des taux d’ARNm de la sérine racémase dans le striatum, l’hippocampe et le cortex des rats, un effet qui devrait induire une augmentation plutôt qu’une diminution des taux de D-sérine. En effet, une administration unique de (R)-cétamine (10 mg/kg, i.p.) a légèrement mais significativement augmenté le rapport D-sérine/L-sérine corticale chez la souris. Par conséquent, une confirmation supplémentaire in vivo des effets de la kétamine et de ses énantiomères sur les niveaux de D-sérine est justifiée.
Il a également été démontré que le DHNK modifie les taux de D-sérine. Singh et al. (2013) ont démontré que l’incubation de cellules PC-12 et 1321N1 avec 5-90 nM de DHNK diminuait les concentrations intracellulaires relatives de D-sérine. Comme le DHNK n’est pas produit dans le cerveau et ne traverse pas la barrière hémato-encéphalique chez les rongeurs traités à la kétamine, la pertinence comportementale des actions de ce métabolite sur les niveaux de D-sérine n’est pas claire.
Les HNK sont également capables de réduire les concentrations intracellulaires de D-sérine dans les cellules PC-12, la (2S,6S)-HNK étant plus puissante que la (2R,6R)-HNK (les IC50 seraient respectivement de 0,18 ± 0,04 et 0,68 ± 0,09 nM). Il est possible que la réduction des taux de D-sérine intracellulaire induite par HNK entraîne également une réduction des taux extracellulaires de cet acide aminé. Cependant, la modulation des taux extracellulaires de D-sérine n’est peut-être pas un déterminant important des effets antidépresseurs des HNK, car, au moins chez la souris, le (2R,6R)-HNK exerce des actions antidépressives plus puissantes que le (2S,6S)-HNK et le DHNK (Sałat et al., 2015). En outre, les études électrophysiologiques n’ont pas permis d’identifier d’effets inhibiteurs des concentrations de ces métabolites pertinentes pour les antidépresseurs sur la fonction des NMDAR, comme le laisserait présager la diminution des taux extracellulaires de D-sérine. En outre, l’administration aiguë de D-sérine induit des réponses comportementales et biochimiques antidépressives de type kétamine chez le rat, ce qui complique encore le rôle fonctionnel possible de la diminution des taux extracellulaires de D-sérine après l’administration de kétamine. Des vérifications supplémentaires, éventuellement en utilisant des cellules dérivées du cerveau humain ou en mesurant les niveaux de D-sérine extracellulaire in vivo après l’administration de kétamine et/ou de ses métabolites, seraient instructives pour déterminer la pertinence fonctionnelle de ces résultats.
B. Canaux à nucléotides cycliques activés par hyperpolarisation
Les canaux HCN sont des canaux cationiques activés par la tension (HCN1-HCN4). L’activation de ces canaux par l’hyperpolarisation de la membrane est facilitée par les nucléotides cycliques, dont l’AMPc. Dans le système nerveux central, les canaux HCN jouent un rôle majeur dans le contrôle de l’excitabilité neuronale, de l’activité synaptique et des oscillations rythmiques.
Il existe un rapport sur les effets inhibiteurs spécifiques des sous-unités de la kétamine (CE50 = 8,2-15,6 μM) sur les canaux hétéromériques HCN1-HCN2 et les courants pacemaker activés par l’hyperpolarisation (Ih ; Chen et al., 2009). Cela pourrait être lié aux actions anesthésiques de la kétamine, car l’anesthésie induite par la kétamine a été significativement supprimée chez les souris knock-out HCN. En outre, la (S)-cétamine s’est avérée plus puissante pour inhiber ces canaux (CE50 = 4,1-7,4 μM) que la kétamine racémique (CE50 = 8,2-15,6 μM ; Chen et al., 2009), ce qui concorde avec la plus grande puissance anesthésique de la (S)-cétamine. En effet, l’hypothèse a été émise que l’inhibition des NMDAR n’est pas le seul mécanisme sous-jacent aux propriétés anesthésiques de la kétamine. D’autres études sont nécessaires pour confirmer le rôle exact de l’inhibition des canaux HCN à cet égard et pour reproduire ces résultats.
Outre un rôle possible dans les propriétés anesthésiques de la kétamine, l’inhibition du canal HCN1 pourrait jouer un rôle dans les actions antidépressives de la kétamine, car la réduction de l’activité HCN1 dans l’hippocampe a été associée à des effets antidépresseurs chez les rongeurs. Il est intéressant de noter que les souris dépourvues du gène HCN1 n’ont pas manifesté de réduction du temps d’immobilité induite par la kétamine dans le test de nage forcée après un traitement chronique à la corticostérone par voie orale. En outre, après l’administration de kétamine, ces souris n’ont pas montré de préférence accrue pour le saccharose ni de diminution de la latence à l’alimentation dans le test d’alimentation supprimée par la nouveauté. Malheureusement, ces résultats ne peuvent pas être interprétés sans ambiguïté comme une preuve que HCN1 médiatise les effets antidépresseurs de la kétamine, car la suppression de HCN1 en elle-même a induit des changements comportementaux de base compatibles avec une réduction du comportement de type dépressif (par exemple, une diminution de l’immobilité dans le test de la nage forcée). En outre, Zhang et al. (2016) ont suggéré que la réduction de la fonction HCN1 par la kétamine était secondaire à l’inhibition des NMDAR présynaptiques. Les auteurs n’ont pas testé l’hypothèse selon laquelle l’inhibition directe de HCN1 par la kétamine, comme l’ont suggéré Petrenko et al. (2014), explique ses effets antidépresseurs. Il n’existe actuellement aucune donnée publiée sur l’activité des métabolites de la kétamine sur la fonction des canaux HCN1 ou l’implication de ces canaux sur les effets comportementaux de ces métabolites.
C. Absorption du GABA et récepteurs GABA
Le principal neurotransmetteur inhibiteur, le GABA, active à la fois les sous-types A et C (GABAA et GABAC) des récepteurs ionotropiques du GABA et le sous-type B (GABAB) des récepteurs métabotropiques du GABA dans le cerveau. Des études électrophysiologiques ont révélé que de fortes concentrations de kétamine potentialisent les courants postsynaptiques inhibiteurs GABAergiques dans les neurones de tranches corticales olfactives de cobaye (300 μM) et de tranches hippocampiques de rat (500 μM). À des concentrations élevées, la kétamine potentialise les récepteurs GABAA activés par le GABA et exprimés de manière ectopique dans les ovocytes de Xénope (365 μM) et les cellules HEK293 (>500 μM ; CE50 = 1,2 mM). Il existe également des preuves d’un effet inhibiteur de la kétamine sur le captage du GABA (Ki = 6,2 ± 1,1 ; IC50 = 50 μM), évalué par le test de liaison au [3H]GABA dans les synaptosomes striataux de rats, ce qui indique que la kétamine pourrait entraîner une augmentation des taux extracellulaires de GABA. En effet, il a été démontré que l’administration intramusculaire de kétamine à des rats augmente la teneur en GABA dans le cerveau et dans les fractions enrichies en synaptosomes du cerveau. Cependant, l’effet de la kétamine sur la captation du GABA n’a pas été constaté à des concentrations cliniquement pertinentes chez la souris (IC50 > 1000 μM).
La pertinence fonctionnelle des actions de la kétamine sur les récepteurs GABAA n’est pas claire car les concentrations de kétamine nécessaires pour modifier l’activité de ces récepteurs sont beaucoup plus élevées que celles obtenues après l’administration de kétamine en milieu clinique pour des effets anesthésiques, analgésiques, anti-inflammatoires et antidépresseurs. Néanmoins, il existe des preuves précliniques des propriétés agonistes et antagonistes de la kétamine sur les récepteurs GABAA. Par exemple, l’administration périphérique de doses infraliminaires de kétamine (0,1 mg/kg, i.p.) combinée à l’agoniste des récepteurs GABAA, le muscimol (0,1 mg/kg, i.p.), a induit une réponse comportementale antidépressive synergique dans le test de nage forcée aiguë (30 minutes après l’injection) chez la souris. En revanche, la perfusion directe de muscimol dans le cortex préfrontal infralimbique a aboli les effets antidépresseurs durables (24 heures après l’injection) de la kétamine sur le comportement des rats, ce qui suggère que l’interaction in vivo entre la kétamine et les récepteurs GABAA pourrait être spécifique à une région du cerveau. En outre, le délire associé à la kétamine survenant à des doses anesthésiques (1-2 mg/kg, perfusion i.v.) est efficacement minimisé par un prétraitement aux benzodiazépines (modulateurs allostériques positifs des récepteurs GABAA). En revanche, Irifune et al. (2000) ont montré que le muscimol et le diazépam, agoniste des récepteurs des benzodiazépines, augmentent l’anesthésie induite par la kétamine, tandis que la bicuculline, antagoniste des récepteurs GABAA, antagonise l’anesthésie induite par la kétamine chez la souris. Enfin, à des doses subanesthésiques, la kétamine ne se lie pas aux récepteurs GABAA dans le cerveau humain, comme l’a montré l’imagerie par tomographie par émission de positons (TEP), et n’altère pas la fonction des récepteurs GABAA à des concentrations pertinentes pour l’anesthésie (c’est-à-dire 10 μM) dans les cellules HEK293 in vitro. Ces résultats indiquent que, au moins à des doses subanesthésiques, la kétamine elle-même pourrait n’affecter qu’indirectement l’activité des récepteurs GABAA pour exercer des effets comportementaux pertinents. En effet, on suppose que les effets dissociatifs/psychotomimétiques induits par la kétamine sont dus au blocage des NMDAR sur les interneurones inhibiteurs GABAergiques, une action qui est supposée désinhiber la neurotransmission excitatrice par une diminution de la libération de GABA et, par conséquent, réduire l’activation des récepteurs GABAA dans les synapses glutamatergiques.
D. Récepteurs cholinergiques
La kétamine se lie aux récepteurs muscariniques et nicotiniques de l’acétylcholine (mAChR et nAChR, respectivement). À ce jour, cinq sous-types de mAChR ont été identifiés (M1-M5). Il s’agit de récepteurs métabotropiques qui signalent principalement, mais pas exclusivement, par l’intermédiaire de Gαi/o (M1, M3 et M5) ou Gαq. La liaison de la kétamine aux sous-types M1, M2 et M3 du mAChR a été décrite. En particulier, l’évaluation du déplacement du [3H]quinuclidinyl benzilate a révélé que la (S)-cétamine a une affinité deux fois plus élevée que la (R)-cétamine pour les mAChR. Dans une étude ultérieure, Hirota et al. (2002) ont démontré que, avec des valeurs Ki d’environ 45, 294 et 246 μM, respectivement, la kétamine déplaçait la liaison de la [3H]N-méthyl scopolamine aux mAChR M1, M2 et M3 exprimés de manière ectopique dans des cellules ovariennes de hamster chinois (CHO). Cependant, les auteurs ont signalé que la kétamine n’avait pas d’effet significatif sur les signaux Ca2+ basaux ou induits par la méthacholine dans les cellules CHO exprimant M1. En revanche, Durieux (1995) a rapporté qu’à des concentrations cliniquement pertinentes, la kétamine inhibait l’activation du mAChR M1 dans les ovocytes de Xenopus (IC50 = 5,7 μM). Ces résultats apparemment divergents pourraient s’expliquer par le fait que les récepteurs exprimés de manière ectopique dans les cellules CHO et les ovocytes peuvent avoir une sensibilité différente. Ces résultats suggèrent la nécessité d’études supplémentaires pour identifier les effets exacts de la kétamine sur les mAChR et la pertinence fonctionnelle de ces effets.
Contrairement aux mAChR métabotropiques, les nAChR sont des récepteurs ionotropiques, qui sont des canaux cationiques non sélectifs activés par le neurotransmetteur acétylcholine. Ces récepteurs sont composés de cinq sous-unités. À ce jour, 10 sous-unités α (α1-α10) et quatre sous-unités β (β1-β4) nAChR ont été clonées, et différentes combinaisons de ces sous-unités donnent naissance à un certain nombre de sous-types nAChR fonctionnels, qui sont exprimés dans les cellules neuronales et non neuronales et possèdent des propriétés pharmacologiques, fonctionnelles et cinétiques spécifiques.
La kétamine agirait comme un bloqueur non compétitif à canal ouvert des sous-types α7, α4β2, α4β4 et α3β4 des nAChR. La concentration de kétamine a bloqué de manière dépendante l’activation induite par l’acétylcholine (1 mM) des nAChRs α4β4 exprimés de manière ectopique dans les ovocytes de Xénope (IC50 = 0,24 ± 0,03 μM), l’inhibition complète étant obtenue avec 10 μM de kétamine (Flood et Krasowski, 2000). De plus, avec des valeurs IC50 de 20 et 50 μM, respectivement, la kétamine a inhibé l’activation induite par l’acétylcholine (1 mM) des nAChRs α7 et α4β2 exprimés de façon ectopique dans les ovocytes. De plus, Moaddel et al. (2013) ont montré que la kétamine inhibe l’activation des nAChRs α3β4 induite par la nicotine (IC50 = 3,1 μM). Étant donné que les concentrations de kétamine allant jusqu’à ∼10 μM se situent dans la plage cliniquement pertinente, certains sous-types de nAChR peuvent sous-tendre les effets de la kétamine in vivo.
Moaddel et al. (2013) ont également démontré qu’à 100 nM, le (R,S)-DHNK réduisait d’environ 60 % l’amplitude des courants de cellules entières induits par l’acétylcholine dans les cellules KXα7R1 qui expriment de manière ectopique les nAChR α7. À la même concentration, les métabolites (2S,6S)-HNK, (2R,6R)-HNK et (R,S)-norkétamine ont également réduit l’amplitude des courants α7 nAChR induits par l’acétylcholine d’environ 54 %, 51 % et 45 %, respectivement. Dans cette étude, les auteurs ont rapporté que le (R,S)-DHNK n’agissait pas comme un bloqueur de canal au niveau des nAChRs α7, car son effet inhibiteur était indépendant du voltage. De plus, le (R,S)-DHNK ne se liait pas au site de liaison agoniste des nAChRs α7, car il ne déplaçait pas la liaison à l’α-bungarotoxine, ce qui suggère que ce métabolite pourrait agir comme un modulateur allostérique négatif.
L’activité antagoniste α7 nAChR des métabolites de la kétamine peut avoir des implications sur l’action antidépressive de la kétamine. Dans ce contexte, il convient de noter que le blocage des nAChR α7 entraîne des effets antidépresseurs dans des modèles de rongeurs. Ainsi, la modulation de l’activité des nAChRs α7 pourrait également être l’un des mécanismes sous-jacents impliqués dans les actions antidépressives de la kétamine, éventuellement par l’intermédiaire de ses métabolites. À l’appui de cette hypothèse, des antagonistes des nAChR font déjà l’objet d’essais cliniques pour le traitement de la dépression.
D’autres sous-types de nAChR sont également sensibles à l’inhibition par des concentrations thérapeutiquement pertinentes de métabolites de la kétamine. Plus précisément, dans les cellules KXα3β4R2 exprimant de manière stable les nAChRs α3β4 de rat, la kétamine et la (R,S)-norkétamine ont été signalées comme inhibant les nAChR α3β4 avec des valeurs IC50 d’environ 3,1 et 9,1 μM, respectivement, tandis que la DHNK, la (2S,6S)- ou la (2R,6R)-HNK n’ont pas eu d’effet significatif sur ces récepteurs (IC50 > 200 μM). On ne peut donc pas exclure la possibilité que les α3β4 nAChRs contribuent également aux effets pharmacologiques de la kétamine.
E. Récepteurs et transporteurs monoaminergiques
Les récepteurs de la dopamine (DA) et de la sérotonine [5-hydroxytryptamine (5-HT)] sont des récepteurs métabotropiques, à l’exception du sous-type 3 du récepteur 5-HT, qui est ionotropique. Cinq sous-types différents de récepteurs DA (D1-5R) et sept sous-types de récepteurs 5-HT (5-HT1-7R) ont été caractérisés dans le système nerveux central. Les transporteurs de neurotransmetteurs régulent l’absorption de DA et de 5-HT à travers les membranes cellulaires/intracellulaires et jouent un rôle clé dans la régulation de la neurotransmission dopaminergique et sérotoninergique.
Bien qu’il existe des preuves que la kétamine puisse agir sur les récepteurs et les transporteurs DA (voir tableau 3), il existe également des preuves contradictoires indiquant que la kétamine ne modifie pas directement la signalisation dopaminergique ; voir également Kokkinou et al. (2018). La kétamine aurait une forte affinité (0,06-1,0 μM) pour les récepteurs D2 (D2R) et agirait comme un agoniste partiel au niveau de ces récepteurs (CE50 = 0,9 ± 0,4 μM). Cette activité agoniste partielle au niveau des D2R contribuerait aux effets psychotomimétiques de la kétamine. Cette hypothèse est également étayée par le fait qu’une diminution de la liaison D2R induite par la kétamine est significativement corrélée aux symptômes liés à la schizophrénie chez l’homme, tels que mesurés par TEP.
Plusieurs études publiées font état d’une diminution de la liaison D2R striatale (mesure indirecte de la libération de DA) induite par la kétamine chez l’homme. Plus précisément, des doses subanesthésiques de kétamine (bolus i.v. de kétamine ; 0,12 mg/kg), suivies d’une perfusion i.v. de 0,65 mg/kg de kétamine pendant 60 minutes (0,29-0,45 μM ; Breier et al., 1998), de 0,5 mg/kg de kétamine pendant 20 minutes (Smith et al., 1998), ou de 0,014 mg/kg par minute de (S)-kétamine pendant 90 minutes (Vollenweider et al., 2000) ont diminué la liaison du D2R striatal. Cependant, des études ultérieures n’ont pas réussi à reproduire ces résultats à l’aide de techniques de tomographie d’émission monophotonique/TEP. En particulier, lorsque la kétamine a été administrée en bolus i.v. (0,12 mg/kg) suivi d’une perfusion i.v. constante de 70 minutes (0,65 mg/kg par heure), entraînant une concentration plasmatique moyenne de 0,59 ± 0,22 μM chez des individus sains, il n’y a pas eu de changement dans la disponibilité des D2R dans les régions striatales du cerveau (Kegeles et al., 2002). De même, la perfusion i.v. de kétamine (perfusion de 66 minutes donnant une concentration plasmatique stable de kétamine de 1,23 ± 0,12 μM) chez des volontaires sains de sexe masculin n’a pas modifié la liaison D2R striatale (Aalto et al., 2002). Conformément à l’absence d’effets directs de doses subanesthésiques de kétamine sur le D2R de la dopamine, les effets psychotomimétiques d’un bolus de 0,23 mg/kg ou d’une perfusion de 0,65 mg/kg par heure de kétamine n’ont pas été bloqués par l’administration d’un antagoniste du D2R chez l’homme. En outre, la kétamine n’a pas modifié la libération de DA accumbale évoquée électriquement et mesurée en temps réel par voltampérométrie cyclique à balayage rapide chez la souris. Contrairement aux études mentionnées précédemment, il a également été rapporté que la kétamine n’a pas d’activité agoniste et antagoniste fonctionnelle sur tous les sous-types de récepteurs DA à des concentrations allant jusqu’à 10 μM.
L’inhibition du transporteur DA par la kétamine dans les cellules HEK293 a également été signalée (Ki = 62,9 μM). Cependant, cet effet ne s’est pas produit à des concentrations allant jusqu’à 10 μM. Comme cet effet n’a été observé qu’à des concentrations relativement élevées, sa pertinence fonctionnelle/clinique reste à déterminer.
La kétamine se lierait également aux récepteurs 5-HT2 avec une affinité de 15 ± 5 μM. Ce résultat pourrait être pertinent pour les effets analgésiques du médicament, car l’antagoniste des récepteurs 5-HT2B/2C, le méthysergide, a inhibé les effets analgésiques de la kétamine chez les rats, ce qui implique la signalisation sérotonergique dans les mécanismes de l’analgésie à la kétamine. En outre, l’administration de kétamine a induit une augmentation des taux extracellulaires de sérotonine (5-HT) dans le cortex préfrontal et le noyau du raphé dorsal des souris.
Chez les primates non humains, il a été démontré que l’administration de kétamine augmentait de manière significative la liaison des récepteurs 5-HT1B de l’accumbal et du pallidum ventral, un effet qui était bloqué par un prétraitement avec l’antagoniste des récepteurs α-amino-3-hydroxy-5-méthyl-4-isoxazolepropionique (AMPA) 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX). Cette découverte peut suggérer que cet effet de la kétamine est impliqué dans ses actions antidépressives, car l’activation des récepteurs AMPA est un mécanisme convergent des actions antidépressives de la kétamine, et l’administration de NBQX abolit les effets de la kétamine dans plusieurs tests animaux d’efficacité antidépressive. Des actions antagonistes de la kétamine sur les récepteurs 5-HT3 ont été rapportées, mais elles se produisent à des concentrations plus élevées que celles cliniquement pertinentes (Ki > 90 μM ; IC50 > 100 μΜ).
Plusieurs études suggèrent que l’augmentation des niveaux de 5-HT est nécessaire pour les effets antidépresseurs de la kétamine chez les rongeurs. Par exemple, les traitements pharmacologiques qui réduisent les niveaux de 5-HT dans le cerveau ont aboli les effets comportementaux antidépresseurs de la kétamine dans le test de nage forcée et dans le test d’alimentation supprimée par la nouveauté chez les rongeurs. En outre, des taux extracellulaires de 5-HT plus élevés étaient positivement corrélés à l’activité antidépressive de la kétamine dans le test de nage forcée chez la souris. La question de savoir si ces effets sont dus à une action directe de la kétamine sur les récepteurs 5-HT n’est pas claire et doit faire l’objet d’une étude plus approfondie. Néanmoins, il existe également des données contradictoires sur les effets de la kétamine sur les récepteurs 5-HT, car même à des concentrations très élevées (1 mM), la kétamine ne modifie que légèrement la liaison du [3H]5-HT ou du [3H]spiroperidol aux récepteurs 5-HT1 ou 5-HT2, respectivement. Il est donc possible que la kétamine interagisse avec le captage de la sérotonine plutôt que de se lier directement aux récepteurs de la sérotonine. En effet, l’administration de doses antidépressives de kétamine (1,5 mg/kg ; perfusion de 40 minutes) à des primates non humains a réduit l’activité des transporteurs de sérotonine (SERT) (Yamamoto et al., 2013), un effet dont on a supposé qu’il reflétait la liaison directe de la kétamine aux SERT pour réguler le recaptage de la 5-HT. Cependant, des travaux in vitro ont indiqué que la kétamine inhibe les SERT à des concentrations allant de 75 à 162 μM (Nishimura et al., 1998), qui sont non seulement supérieures aux concentrations pertinentes pour les antidépresseurs, mais aussi bien supérieures aux concentrations anesthésiques cliniques de la kétamine. À des concentrations pertinentes pour les antidépresseurs, la kétamine n’a pas d’effet agoniste ou antagoniste sur les SERT. Par conséquent, les preuves in vivo de l’inhibition de la recapture de la sérotonine induite par la kétamine pourraient être attribuées aux interactions indirectes de la kétamine avec le système sérotoninergique, du moins à des concentrations subanesthésiques.
Enfin, bien qu’il existe des preuves que la kétamine agit comme un inhibiteur de la recapture au niveau des transporteurs de la norépinéphrine (NET ; Ki = 66,8 μM), la constante d’inhibition indique que la kétamine ne modulerait pas la fonction des NET à des concentrations cliniquement pertinentes (jusqu’à 10 μΜ). On note également l’absence d’activité fonctionnelle de concentrations cliniquement pertinentes de kétamine (jusqu’à 10 μΜ) sur les NET.
À ce jour, il n’existe qu’une seule étude publiée évaluant l’effet des métabolites de la kétamine sur les récepteurs et les transporteurs monoaminergiques. À des concentrations allant jusqu’à 10 μM, il n’y a pas eu d’activité agoniste ou antagoniste de la (S)-norkétamine, de la (R)-norkétamine, de la (S)-déhydronorkétamine, de la (R)-déhydronorkétamine, de la (2S,6S)-HNK ou de la (2R,6R)-HNK sur les récepteurs D1, D2, D3, D4 ou D5, le transporteur DA, la NET ou la SERT. Bien que ces résultats suggèrent que les effets directs des métabolites de la kétamine sur les récepteurs DA ou les transporteurs monoaminergiques n’expliquent pas les actions antidépressives de la kétamine, il n’est pas exclu qu’à des concentrations plus élevées, les métabolites de la kétamine interagissent avec ces récepteurs et transporteurs et en modifient directement ou indirectement l’activité.
F. Récepteurs opioïdes
Les récepteurs opioïdes sont exprimés dans tout le système nerveux central ainsi que dans les tissus périphériques. Ces récepteurs sont couplés aux protéines G et sont classés en trois sous-types (récepteurs opioïdes μ-, δ- et κ). L’une des principales fonctions de l’activation des récepteurs opioïdes est d’inhiber la transmission des stimuli nociceptifs, ce qui entraîne une analgésie.
On a signalé que la kétamine activait les récepteurs opioïdes recombinants humains μ-, κ- et δ- exprimés dans des cellules CHO (Ki = 42,1, 28,1 et 272 μΜ, respectivement). Il a été démontré que la (S)-cétamine avait une affinité plus élevée que la (R)-cétamine pour se lier aux récepteurs opioïdes. En particulier, les affinités de la (S)-cétamine varient de 11 à 29 μΜ pour les récepteurs μ-, de 25 à 28 μΜ pour les récepteurs κ-, et de 130 à 205 μΜ pour les récepteurs δ-opioïdes. En revanche, la (R)-cétamine a des affinités allant de 28 à 84 μΜ pour les récepteurs μ-, de 60 à 100 μΜ pour les récepteurs κ- et de 130 à 286 μΜ pour les récepteurs δ-opioïdes (Hustveit et al., 1995 ; Hirota et al., 1999). À l’instar de ces résultats, la (S)-cétamine (CI50 = 23,0 ± 1,2 μM) a montré une plus grande capacité à se lier et à déplacer le ligand opioïde non spécifique [3H]dihydromorphine par rapport à la kétamine racémique (CI50 = 16,3 ± 7,4 μM) et à la (R)-cétamine (CI50 = 45,5 ± 3,2 μM).
On suppose que les actions de la kétamine sur les récepteurs opioïdes sont impliquées dans ses effets analgésiques, et ces résultats sont cohérents avec la puissance plus élevée de la (S)-kétamine dans les mesures de l’antinociception par rapport à la (R)-kétamine. Cependant, le rôle exact des récepteurs opioïdes dans la médiation de ces effets n’est pas clair.
Bien que les actions analgésiques de la kétamine soient bloquées par l’administration intracérébroventriculaire (c’est-à-dire l’exposition directe du cerveau) d’antagonistes des récepteurs opioïdes μ- et δ-, mais pas κ- chez la souris, l’inhibition globale des récepteurs opioïdes obtenue par l’administration systémique de naloxone n’empêche pas l’analgésie induite par la kétamine chez l’homme. Ces résultats apparemment opposés indiquent que la kétamine pourrait induire une analgésie par le biais d’une interaction indirecte avec le système opioïde, ou qu’elle pourrait exercer une action spécifique au sous-type de récepteur opioïde. À l’appui d’un tel effet spécifique au sous-type, des données in vivo indiquent que la kétamine pourrait être un agoniste des récepteurs κ-opioïdes et un antagoniste des récepteurs μ-opioïdes. En particulier, la micro-injection de kétamine dans la région grise périaqueducale, qui contient des récepteurs μ- mais pas κ-opioïdes, n’a pas eu d’effets antinociceptifs, mais a bloqué les effets de l’agoniste des récepteurs μ-opioïdes, la morphine, dans la même région. Cela pourrait expliquer l’incapacité de la naloxone à bloquer l’analgésie induite par la kétamine et plaide en faveur d’un rôle spécifique des récepteurs opioïdes dans l’analgésie induite par la kétamine. Toutefois, il convient de noter que ces résultats vont à l’encontre de ceux de Pacheco Dda et al. (2014), comme nous l’avons vu plus haut. En outre, il a été rapporté que des doses sous-effectives de kétamine n’antagonisent que partiellement les actions cataleptiques de la morphine chez le rat ; cependant, une combinaison de doses sous-effectives de kétamine et de morphine a induit une catalepsie dans les mêmes conditions expérimentales. Dans l’ensemble, ces résultats impliquent le système opioïde dans les effets analgésiques de la kétamine, mais d’autres études sont nécessaires pour clarifier les mécanismes exacts. Les interactions entre la kétamine et le système opioïde peuvent être plus pertinentes dans la douleur chronique, où la kétamine réduit la tolérance aux opioïdes. En particulier, en agissant sur la voie en aval de la kinase régulée par le signal extracellulaire 1/2, la kétamine (10 μΜ) a aboli la désensibilisation induite par les récepteurs μ-opioïdes in vitro.
L’activation des récepteurs κ-opioïdes par la kétamine (CE50 = 29 μΜ) serait impliquée dans les effets de la kétamine sur l’attention et la perception visuelle chez les rats évalués dans la tâche de temps de réaction en série à cinq choix. Le prétraitement par un antagoniste sélectif des récepteurs κ-opioïdes a bloqué la capacité de la kétamine (20 mg/kg) à altérer l’attention et la perception visuelle (Nemeth et al., 2010). En revanche, chez des volontaires sains, l’inhibition des récepteurs opioïdes par la naloxone a augmenté les effets dissociatifs et les déficits cognitifs induits par une administration de kétamine à un seuil inférieur (bolus de 0,23 mg/kg pendant 1 minute, suivi d’une perfusion de 0,58 mg/kg de kétamine pendant 60 minutes), ce qui pourrait indiquer une activité antagoniste de faibles doses de kétamine sur les récepteurs opioïdes in vivo. Néanmoins, des travaux supplémentaires sont nécessaires pour établir le rôle exact de la modulation des récepteurs opioïdes dans la médiation des effets secondaires de la kétamine. Il n’existe actuellement aucune étude publiée évaluant les effets des métabolites de la kétamine sur les récepteurs opioïdes. Cependant, des données récentes indiquent que le (2R,6R;2S,6S)-HNK (à 10 et/ou 30 mg/kg, s.c.) ne possède pas de propriétés antinociceptives et ne modifie pas la tolérance aux opioïdes (morphine) chez le rat, ce qui indique une absence possible d’interactions entre ce métabolite et le système des récepteurs opioïdes. Notamment, la norkétamine, à l’instar de la kétamine, atténue la tolérance à la morphine.
G. Récepteurs sigma
Un autre site d’action potentiel de la kétamine est le récepteur sigma. Les récepteurs sigma sont classés en deux sous-types différents, les récepteurs sigma I et II (σ1R et σ2R, respectivement ; Bowen et al., 1989). Bien que l’existence d’un troisième sous-type ait été suggérée (σ3R), il n’a pas été entièrement défini.
Comme le montre le tableau 3, la liaison de la kétamine a été décrite à la fois au niveau du σ1R et du σ2R. La première preuve de la liaison directe de la kétamine aux récepteurs sigma a été apportée par Klepstad et al. (1990), qui ont démontré que la (R)-kétamine a une puissance de CI50 = ∼15 μM pour se lier aux récepteurs sigma, tandis que la (S)-kétamine a une CI50 de ∼100 μM. Bien que ces résultats soient qualitatifs et non absolument quantitatifs en raison des différentes régions du cerveau utilisées pour les études de liaison, Hustveit et al. (1995) ont confirmé ces premiers résultats en montrant que la (R)-cétamine a une affinité de 19 μM pour les récepteurs sigma, tandis que la (S)-cétamine présente une affinité de liaison ∼15 fois plus faible pour ces récepteurs (131 μM). Robson et al. (2012) ont rapporté que la (R,S)-cétamine a une affinité préférentielle pour le σ2R (Ki = 26,3 μΜ) par rapport au σ1R (Ki = 139,6 μΜ). À l’appui de cette liaison in vivo, des études menées sur des primates non humains ont montré une liaison compétitive entre la kétamine et le [11C]SA5845, un traceur TEP des récepteurs sigma (Kortekaas et al., 2008).
Les récepteurs sigma sont des cibles prometteuses pour le traitement antidépresseur, et l’activation de ces récepteurs induit des réponses comportementales antidépressives chez les animaux et les humains. On peut donc supposer que l’action de la kétamine sur les récepteurs sigma est impliquée dans les mécanismes qui sous-tendent ses réponses antidépressives, ce qui est cohérent avec l’affinité de liaison plus élevée de la (R)-cétamine pour ces récepteurs et ses effets antidépresseurs plus puissants que ceux de la (S)-cétamine dans les modèles de rongeurs.
Bien que l’administration d’antagonistes σ1R et σ2R n’ait pas atténué les effets antidépresseurs de la kétamine sur le comportement des souris, l’administration d’un antagoniste sélectif du σ1R a bloqué les effets potentialisateurs de la kétamine sur l’excroissance neuritique induite par le facteur de croissance nerveuse, et donc la modulation des voies liées à la neuroplasticité qui sont impliquées dans les effets antidépresseurs du médicament (voir Kavalali et Monteggia (2012), Gerhard et al. (2016)). Ces résultats pourraient indiquer que les actions de la kétamine sur les récepteurs sigma pourraient être impliquées dans les effets liés à la neuroplasticité du médicament, et donc indirectement dans ses actions antidépressives. Il n’existe actuellement aucune preuve de l’activité des métabolites de la kétamine sur les récepteurs sigma.
H. Canaux sodiques à potentiel d’action
Les canaux ioniques voltage-dépendants sont parmi les premiers canaux ioniques identifiés et sont impliqués dans la génération de potentiels d’action. Les anesthésiques locaux induisent généralement une inhibition concentration-dépendante de l’activité des canaux sodiques, en se liant à des sites situés dans le pore du canal. Alors que la kétamine s’est avérée agir comme un anesthésique local dans la pratique vétérinaire et chez les patients humains, il existe actuellement des preuves contradictoires concernant les effets de la kétamine sur l’activité du canal sodique voltage-gated.
Dans des myocytes ventriculaires isolés de cobaye, la kétamine, à des concentrations allant de 30 à 300 μΜ, a induit une inhibition tonique des courants des canaux sodiques en fonction de la concentration, mais pas de l’utilisation (16%-36% d’inhibition ; Hara et al., 1998b). En revanche, la kétamine a induit un blocage tonique (CI50 = 866,2 ± 34,7 μΜ) et phasique (CI50 = 314,8 μΜ) des canaux sodiques dans les neurones du ganglion de la racine dorsale du rat. En outre, la kétamine bloquerait les canaux sodiques voltage-dépendants dans les ovocytes de Xenopus (inhibition tonique : IC50 = 800 μΜ ; inhibition phasique : IC50 = 2,3 mM). En outre, bien que Benoît (1995) n’ait démontré aucun effet inhibiteur de la kétamine sur les courants des canaux sodiques nodaux des fibres nerveuses myélinisées, d’autres ont rapporté un blocage allant jusqu’à 71,1 % de la conductance des canaux sodiques par la kétamine (ED50 = 1,1 mM). Une étude évaluant les effets de la (S)- et de la (R)-cétamine sur l’activité des canaux sodiques a montré que la (S)-cétamine inhibe les canaux sodiques voltage-gated avec une puissance apparente de 240 ± 60 et 59 ± 10 μΜ dans les isoformes neuronales et musculaires squelettiques, respectivement. La capacité de la (R)-cétamine à bloquer les canaux sodiques était plus faible (IC50 = 333 ± 93 et 181 ± 49 μΜ dans les isoformes neuronales et musculaires squelettiques, respectivement) que celle de la (S)-cétamine. De même, Schnoebel et al. (2005) ont montré un blocage tonique stéréosélectif des courants sodiques voltage-dépendants, la (S)-cétamine étant plus puissante que la (R)-cétamine (IC50 = 128 et 269 μΜ, respectivement). Ces résultats indiquent que la (S)-cétamine est un anesthésique local plus efficace que la (R)-cétamine.
Ces divergences dans les effets de la kétamine sur l’activité des canaux sodiques pourraient être dues à des différences dans les populations cellulaires et les procédures expérimentales utilisées. Dans l’ensemble, ces résultats confirment l’effet inhibiteur de la kétamine sur les canaux sodiques, caractéristique des anesthésiques locaux. Ces effets se produisent à des concentrations bien supérieures aux niveaux de kétamine circulante pertinents pour l’anesthésie générale, mais pourraient éventuellement être pertinents pour l’anesthésie locale à la kétamine.
I. Canaux calciques voltage-dépendants de type L
La famille des canaux calciques voltage-dépendants de type L (VDCC) se compose de quatre membres différents appelés Cav1.1-Cav1.4. Cav1.2 est le principal VDCC exprimé dans le cerveau des mammifères. L’antagonisme des VDCC de type L par la kétamine a été rapporté dans une large gamme de concentrations. Dans les cellules musculaires lisses de la trachée porcine, par exemple, la kétamine a bloqué les VDCC avec une CI50 de 1 mM. Dans les cellules lisses de la veine porte du lapin, 1 mM de kétamine a complètement inhibé les courants VDCC. Enfin, dans les cellules auriculaires simples de grenouille, la kétamine a inhibé les courants VDCC avec une CI50 de 9,2 μΜ. Cet effet n’était pas dépendant de l’utilisation et la kétamine n’a pas agi comme un bloqueur de canaux ouverts. Une inhibition tonique non dépendante de l’utilisation des VDCC a également été signalée dans des myocytes ventriculaires isolés de cobaye, dans lesquels 30-300 μΜ ont induit une inhibition de 26%-53%. Les divergences dans les concentrations rapportées auxquelles la kétamine inhibe les VDCC peuvent être dues à des différences d’espèces, de types de cellules ou de préparations expérimentales (par exemple, différences dans les solutions de bain).
Malgré les preuves de l’inhibition des VDCC par la kétamine, une étude in vitro a montré que l’activation des récepteurs AMPA et l’augmentation subséquente de la libération du facteur neurotrophique dérivé du cerveau et de l’activation du complexe 1 de la cible mécaniste de la rapamycine – actions qui sont supposées sous-tendre les propriétés antidépressives de la kétamine – nécessitent l’activation des VDCC. En outre, les effets antidépresseurs de la kétamine chez la souris sont bloqués par un prétraitement avec des antagonistes des canaux calciques de type L. Ces résultats ne sont pas en accord avec l’activité inhibitrice de la kétamine au niveau de ces canaux, telle que décrite précédemment. Toutefois, il convient de noter que ces études ont été réalisées sur des cellules dérivées de tissus périphériques (par exemple, le cœur, la trachée) et que les effets de la kétamine sur les VDCC dans les cellules dérivées du cerveau à des concentrations pertinentes pour les antidépresseurs n’ont pas été rapportés à notre connaissance. Outre le rôle possible de l’inhibition des canaux calciques induite par la kétamine dans la médiation des actions antidépressives du médicament, le blocage des canaux calciques a également été supposé être impliqué dans les effets psychoactifs/psychotomimétiques de la kétamine.
IV. Conclusions
La kétamine est utilisée en clinique comme anesthésique depuis les années 1970. Cependant, de nouvelles indications (par exemple, la douleur chronique et la dépression) et de nouveaux mécanismes d’action (par exemple, les métabolites HNK) sont encore en train d’émerger. Son utilisation comme anesthésique, analgésique, anti-inflammatoire et antidépresseur a relancé la recherche pour comprendre les mécanismes neurobiologiques qui sous-tendent ces effets de la kétamine, ainsi que de ses métabolites, et plusieurs cibles moléculaires et cellulaires ont été identifiées à ce jour.
La kétamine est généralement utilisée en clinique (et en recherche préclinique) sous la forme d’un mélange racémique, la (R-S)-kétamine. Elle est largement et rapidement métabolisée in vivo, ce qui entraîne la formation de norkétamine et de HK, suivie de la production de métabolites secondaires, le DHNK et les HNK. Bien que la kétamine, la norkétamine et les HNK traversent facilement la barrière hémato-encéphalique, le DHNK ne semble pas atteindre des concentrations cérébrales pharmacologiquement actives, du moins dans le cerveau de la souris. La kétamine et, dans une moindre mesure, la norkétamine, sont des antagonistes non compétitifs du canal ionique NMDAR. Les DHNK et les HNK semblent toutefois avoir un pouvoir d’inhibition du NMDAR beaucoup plus faible, voire nul.
Outre le NMDAR, nous avons examiné les résultats étayant les actions de la kétamine sur un certain nombre de récepteurs et de canaux ioniques, notamment les récepteurs DA, 5-HT, opioïdes, cholinergiques, sigma et GABAA, ainsi que les transporteurs de monoamines et les VDCC de type HCN, sodium et L. Les effets de la kétamine sur les récepteurs et les canaux ioniques sont bien connus et bien documentés. Les effets anesthésiques bien caractérisés de la kétamine sont principalement attribués à l’inhibition des NMDAR. Cependant, il existe des preuves que l’inhibition des canaux HCN pourrait également être impliquée dans les propriétés anesthésiques de la kétamine. De même, des données indiquent que l’inhibition des NMDAR joue un rôle dans les actions analgésiques de la kétamine, avec la possibilité que les récepteurs opioïdes jouent également un rôle.
A. La (R)- et la (S)-kétamine en tant qu’antidépresseurs
Une grande attention a été accordée au NMDAR en tant que cible pharmacologique principale responsable des actions antidépressives de la kétamine. Cependant, contrairement à cette prédiction, des études cliniques ont suggéré une efficacité antidépressive nulle ou modeste de certains antagonistes NMDAR alternatifs. À ce jour, ces médicaments n’ont pas l’efficacité antidépressive robuste, rapide ou soutenue de la kétamine, et certains (par exemple, la mémantine) se sont révélés cliniquement inefficaces dans de nombreuses études. En outre, Hashimoto et ses collègues ont rapporté pour la première fois des actions antidépressives supérieures et plus durables de la (R)-cétamine par rapport à la (S)-cétamine dans des modèles de rongeurs. Ces résultats ont également été confirmés par Zanos et al. (2016), qui ont montré que les effets comportementaux antidépresseurs de la (S)-cétamine ne deviennent apparents qu’à des doses plus élevées que ceux de la (R)-cétamine. La puissance accrue de la (R)-cétamine ne semble pas être liée à une réponse à la dose en forme de U des médicaments, car elle s’est avérée plus puissante que la (S)-cétamine avec une gamme de doses allant jusqu’à 30 fois dans de multiples tests d’efficacité antidépressive chez la souris. L’administration de doses égales de (R)-cétamine et de (S)-cétamine n’a pas produit de niveaux différents de ces énantiomères dans le cerveau des rongeurs, ce qui indique que l’augmentation de la puissance antidépressive de la (R)-cétamine dans les modèles de rongeurs n’est pas due à des niveaux plus élevés du médicament dans le cerveau. Ces données précliniques sur les rongeurs indiquent que la (R)-cétamine est un antidépresseur plus puissant que l’énantiomère (S)-cétamine, ce qui suggère qu’il est peu probable que la kétamine exerce ses effets antidépresseurs uniquement par l’inhibition du NMDAR. Néanmoins, nous notons que des études précliniques sur les rongeurs ont également indiqué des actions comportementales antidépressives à action rapide de la (S)-cétamine chez les souris.
La découverte que la kétamine exerce une action antidépressive rapide, dans les heures qui suivent son administration, a concentré les efforts de recherche sur la compréhension de ce phénomène. Cette découverte et l’élucidation des mécanismes pertinents impliqués pourraient révolutionner le traitement de la dépression, étant donné que les antidépresseurs actuellement approuvés mettent des semaines, voire des mois, à exercer pleinement leurs effets antidépresseurs, et que de nombreux patients souffrant de troubles dépressifs majeurs sont résistants aux pharmacothérapies antidépressives classiques. Comme pour la kétamine racémique, des études cliniques menées sur des patients déprimés ont montré qu’une perfusion i.v. de 40 minutes de (S)-kétamine exerce des effets antidépresseurs rapides (dans les deux heures suivant l’administration). En outre, des effets antidépresseurs dose-dépendants de la (S)-cétamine administrée par voie intranasale (dose de 28 à 84 mg, deux fois par semaine pendant un total de deux semaines) ont été rapportés chez des patients déprimés résistants au traitement et sous traitement antidépresseur classique par voie orale. La (S)-cétamine fait actuellement l’objet d’essais cliniques de phase III en tant qu’antidépresseur.
Si, dans la dépression humaine, comme c’est le cas dans les modèles murins, la (R)-cétamine a une puissance supérieure à la (S)-cétamine, cela présenterait des avantages compte tenu de ses effets secondaires moindres dus à une inhibition moins puissante du NMDAR (voir la section Récepteurs du N-Méthyl-D-Aspartate). Cependant, il n’existe actuellement aucune étude clinique publiée évaluant l’efficacité antidépressive de la (R)-cétamine chez les patients déprimés. En outre, il n’existe aucune étude clinique publiée comparant directement les actions antidépressives des énantiomères (S)- et (R)-cétamine, ou comparant les actions de l’un ou l’autre des énantiomères au mélange racémique.
B. Utilité des métabolites de l’hydroxynorkétamine de la kétamine comme traitement médicamenteux
Il a été rapporté que le métabolisme de la kétamine est nécessaire pour qu’elle agisse pleinement comme antidépresseur chez la souris. Les métabolites HNK spécifiques de la kétamine, (2S,6S)-HNK et (2R,6R)-HNK, dérivés du métabolisme de la (S)-cétamine et de la (R)-cétamine, respectivement, ne se lient pas au NMDAR et ne l’inhibent pas fonctionnellement à des concentrations pertinentes pour les antidépresseurs, mais exercent des effets comportementaux antidépresseurs similaires à ceux observés après l’administration de la kétamine elle-même. Ces résultats remettent encore plus en question l’hypothèse de l’inhibition du NMDAR dans les actions antidépressives de la kétamine. En outre, la (2R,6R)-HNK exerce des effets antidépresseurs sans la dissociation sensorielle, l’ataxie et le risque d’abus de la kétamine dans les essais sur les animaux. En effet, les effets secondaires psychoactifs de la kétamine, notamment la dissociation et les modifications de la perception sensorielle, ainsi que son potentiel d’abus, ont été attribués aux effets d’inhibition des NMDAR de la kétamine.
Les récentes découvertes selon lesquelles les métabolites de la kétamine sont impliqués dans les actions antidépressives de la kétamine suggèrent l’utilisation possible de ces métabolites dans le traitement de la dépression et ouvrent de nouvelles voies pour l’étude de leur rôle dans d’autres troubles cérébraux. Comme nous l’avons évoqué précédemment, les métabolites HNK peuvent contribuer aux effets cliniques des doses subanesthésiques de kétamine, peut-être en raison de leurs actions directes sur les nAChR, indirectes sur la D-sérine, ou d’autres cibles qui n’ont pas encore été identifiées. Ces travaux peuvent fournir un cadre pour un nouveau paradigme du métabolite de la kétamine, qui postule des effets cliniquement pertinents dépendant de la conversion métabolique de la kétamine, mais qui n’implique pas l’inhibition des NMDAR. Néanmoins, de futures études précliniques sont nécessaires pour étayer l’affirmation selon laquelle l’inhibition des NMDAR n’est pas nécessaire à l’efficacité des métabolites de la kétamine en tant qu’antidépresseurs à action rapide et pour identifier le mécanisme d’action sous-jacent de ces métabolites. Il reste également à déterminer si les métabolites de la kétamine jouent un rôle dans les actions anti-inflammatoires ou analgésiques de la kétamine.
C. Orientations futures
L’identification des cibles responsables des différents effets comportementaux de la kétamine, et potentiellement de ses métabolites, est essentielle pour le développement de nouvelles pharmacothérapies qui n’auront pas les effets secondaires de la kétamine, y compris les effets psychotomimétiques, les modifications de la perception sensorielle et le potentiel d’abus. Outre les études récemment achevées sur des modèles précliniques de dépression, des études sur les métabolites de la kétamine dans des modèles animaux de douleur, d’inflammation, de dépression et de suicidalité sont essentielles pour mieux comprendre leur potentiel thérapeutique. L’utilisation clinique élargie de la kétamine racémique, de la (S)-cétamine, de la (R)-cétamine et des métabolites potentiellement clés [c’est-à-dire (2R,6R)-HNK] représente une opportunité importante de définir de nouvelles thérapies pour des conditions médicales non satisfaites et de mieux définir les relations pharmacologie-phénotype. Notamment, l’exploration clinique de ces agents est possible compte tenu des connaissances historiques sur la sécurité des formes racémiques et énantio-pures de la kétamine (et donc de ses métabolites) lorsqu’elles sont administrées de manière aiguë. Ainsi, les investissements dans les voies d’administration alternatives, les stratégies de dosage et l’évaluation des associations médicamenteuses ne sont pas excessivement lourds. En outre, l’étendue des indications potentielles, associée aux multiples cibles pharmacologiques de la kétamine, offre une fenêtre de compréhension sans précédent sur les nouveaux mécanismes des futures interventions thérapeutiques. Compte tenu de ces facteurs, il est clair que la compréhension globale de la pharmacologie de la kétamine et de ses métabolites offre des possibilités inestimables en matière de recherche fondamentale et translationnelle et de soins cliniques.
