
Sigel, E., & Ernst, M. (2018). The benzodiazepine binding sites of GABAA receptors. Trends in pharmacological sciences, 39(7), 659-671.
Points forts
De multiples sites non canoniques d’action des benzodiazépines, y compris des sites structurellement non homologues dans le domaine transmembranaire, ont été décrits.
Les récepteurs GABAA extrasynaptiques réagissant aux ligands du chimiotype des benzodiazépines sont des cibles médicamenteuses précieuses.
Les approches pharmacogénétiques permettent de démêler la fonction physiologique des sous-unités individuelles des récepteurs.
Les modulateurs allostériques négatifs agissant sélectivement sur des sites de liaison spécifiques des benzodiazépines peuvent, contrairement aux attentes, représenter des médicaments utiles, comme le montre l’exemple du basmisanil.
Les animaux transgéniques ouvrent la voie à de nouvelles indications thérapeutiques potentielles d’une nouvelle génération de benzodiazépines à sélectivité améliorée.
Les méthodes optogénétiques ont commencé à être appliquées à la recherche sur les récepteurs GABAA et peuvent apporter des informations utiles.
L’activité quotidienne repose sur un équilibre subtil entre les systèmes neuronaux excitateurs et inhibiteurs. Les principaux acteurs de l’inhibition neuronale sont les récepteurs GABAA synaptiques et extrasynaptiques. Les benzodiazépines sont des médicaments populaires qui agissent comme des modulateurs allostériques positifs d’un sous-ensemble de ces récepteurs. Les benzodiazépines ont des effets sédatifs, hypnotiques, myorelaxants et anticonvulsifs, et présentent un risque de surdosage exceptionnellement faible. La découverte d’un grand nombre de sous-types de récepteurs GABAA a fait naître l’espoir d’une séparation claire de ce spectre d’actions. Nous examinons ici dans quelle mesure cette séparation a été réalisée et nous décrivons les progrès récents dans la découverte de nouveaux ligands pour les sites de liaison canoniques et non canoniques.
Les benzodiazépines : passé, présent et futur.
Les ligands du site de liaison à haute affinité des benzodiazépines (voir glossaire) sont des médicaments qui ont connu un immense succès au cours des dernières décennies. Contrairement à de nombreux autres médicaments, ils sont pratiquement exempts de toxicité aiguë et chronique. Pour le traitement des troubles du sommeil, ce sont les médicaments de choix, et ils sont souvent utilisés dans les troubles anxieux. Ils sont également utilisés comme sédatifs en anesthésie et en psychiatrie, ainsi que comme relaxants musculaires à action centrale. Pourquoi ces médicaments présentent-ils encore un intérêt scientifique 57 ans après leur mise sur le marché ? Ce type de médicament a-t-il un avenir ? Cette revue explique pourquoi la réponse est toujours oui. Les benzodiazépines exercent leurs effets par l’intermédiaire des récepteurs GABA-A qui répondent au neurotransmetteur GABA. Les médicaments ciblant le site des benzodiazépines, tels que le diazépam ou les drogues Z (encadré 1), se lient avec une grande affinité à certains sous-types de récepteurs GABA-A et avec une faible affinité à différents sites dans la plupart des autres sous-types de récepteurs. La compréhension de ces sous-types de récepteurs offre de nombreuses possibilités de séparer les effets pléiotropes des benzodiazépines qui comprennent la sédation, l’hypnose, l’anxiolyse et la relaxation musculaire. Actuellement, de nouvelles applications des benzodiazépines en neuropsychiatrie sont à l’étude, par exemple pour le renforcement cognitif, le soulagement de la douleur et éventuellement le traitement de la dépression. En outre, les ligands des sites de liaison à haute affinité des benzodiazépines sont des molécules traceuses précieuses pour les méthodes d’imagerie basées sur les ligands, telles que la tomographie par émission de positrons (TEP) et la tomographie par émission monophotonique (TEMP). Les développements récents en matière de découverte de ligands, de caractérisation des sites de liaison et de cartographie des effets pharmacologiques sur des sous-types de récepteurs spécifiques sont passés en revue ici. Nous commençons par examiner les propriétés moléculaires des récepteurs GABA-A.
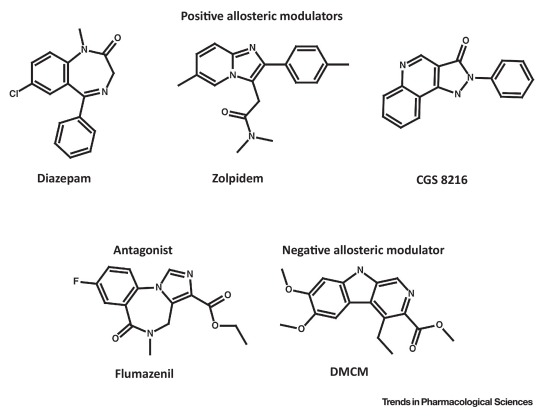
| Encadré 1 Ligands des sites de liaison des benzodiazépines. Alors que les premiers “médicaments benzodiazépines” étaient des 1,4-benzodiazépines (comme le diazépam, Figure I), de nombreux ligands du site de liaison à haute affinité des benzodiazépines qui ont été développés par la suite ont une structure autre que celle des benzodiazépines. Parmi les chimiotypes les plus importants, on trouve les β-carbolines (comme le DMCM et l’abécarnil), le groupe hétérogène des drogues Z (comme le zolpidem, le zaleplon et la zopiclone), et les pyrazoloquinolinones (comme le CGS 8216). La plupart des chimiotypes comprennent des modulateurs allostériques positifs, des modulateurs nuls (ou silencieux) et des modulateurs allostériques négatifs (encadré 3). Les modulateurs allostériques positifs non sélectifs sont anticonvulsivants, sédatifs-hypnotiques et anxiolytiques, tandis que les modulateurs allostériques négatifs non sélectifs sont pro-convulsivants et anxiogènes. Les modulateurs nuls ou silencieux antagonisent ces effets in vivo, comme le flumazénil, antidote des benzodiazépines. |
Les récepteurs GABA-A sont des sites d’action des benzodiazépines.
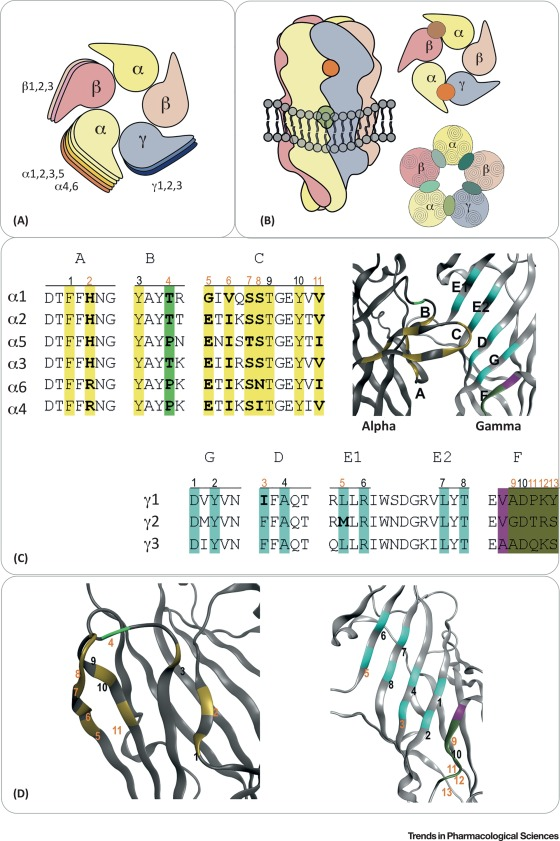
Le site de liaison à haute affinité pour les benzodiazépines a été isolé pour la première fois dans le cerveau d’un bovin et a été décrit comme un complexe protéique avec deux sous-unités appelées α et β [7]. Ce complexe protéique s’est avéré identique au récepteur GABA-A. Le clonage et l’expression de ces deux sous-unités ont confirmé l’identité du complexe protéique. Il a rapidement été reconnu qu’il existait de multiples isoformes de sous-unités et, à ce jour, des sous-unités appelées α1-6, β1-3, γ1-3, ρ1-3, δ, ε, θ, et π ont été identifiées chez les mammifères (Encadré 2). Une à cinq isoformes de ces sous-unités sont assemblées en un complexe pentamérique en forme d’anneau avec un canal central sélectif des ions chlorure et bicarbonate. La fixation du neurotransmetteur GABA ouvre ce canal et les benzodiazépines modulent cette ouverture. L’isoforme de la sous-unité principale se compose de α1, β2 et γ2, disposés α1γ2β2α1β2 dans le sens inverse des aiguilles d’une montre, vu de l’extérieur de la cellule. Les deux sites de liaison pour l’agoniste GABA sont situés aux interfaces des sous-unités extracellulaires β2/α1, et le site de liaison à haute affinité pour les benzodiazépines est situé à une position étroitement homologue à l’interface α1/γ2 de ce récepteur. Il peut exister des dizaines d’autres sous-types de récepteurs GABA-A (encadrés 2,3). Même les récepteurs ayant la même composition en sous-unités peuvent s’assembler en différentes espèces de récepteurs. Il a été démontré que différents compartiments subcellulaires des neurones, tels que les densités postsynaptiques, les régions dendritiques périsynaptiques et les somates cellulaires, contiennent des sous-types de récepteurs distincts.
| Encadré 2 Sous-types de la famille des récepteurs GABA-A Il est généralement admis, avec l’appui de la plupart des preuves expérimentales, que la majorité des récepteurs GABAA dans le cerveau des mammifères adultes sont des pentamères avec une composition de deux sous-unités α1, deux sous-unités β2 (ou deux sous-unités β3) et une sous-unité γ2, à savoir (α1)2(β2 ou β3)2(γ2)1. Pour une grande partie des récepteurs qui contiennent une isoforme de sous-unité α non α1 (c’est-à-dire, α2 à α6), il a été démontré qu’ils contiennent souvent aussi l’une des autres isoformes α, par exemple les récepteurs α1α6βxγ2 et α1α6βxδ dans le cervelet ou les récepteurs contenant α1α2- et α2α3 dans la moelle épinière. En général, pour les récepteurs contenant l’une des six isoformes α et l’une des trois isoformes γ, on pense que la stœchiométrie générale des sous-unités est préservée, à savoir (α)2(β)2(γ)1. La disposition de ces récepteurs est α1γ2β2α1β2 dans le sens inverse des aiguilles d’une montre (vu de l’extérieur de la cellule), ce qui a été vérifié expérimentalement pour les récepteurs α1β(2 ou 3)γ2 (Figure 1). Les récepteurs contenant les sous-unités γ1 ou γ3 sont supposés être disposés comme ceux contenant la sous-unité γ2. La composition et la disposition des récepteurs contenant la sous-unité δ sont moins claires et ont fait l’objet de débats controversés. Il a été démontré que la sous-unité δ s’assemblait avec les sous-unités α4, α6 et α1, ainsi qu’avec toute sous-unité β. La majorité des auteurs favorisent une stœchiométrie (α)2(β)2(δ)1, mais l’arrangement est moins bien établi, et différents groupes utilisant différentes techniques (par exemple, microscopie à force atomique, marquage par photo-affinité des sous-unités voisines, récepteurs recombinants concaténés) ont proposé différents arrangements et un assemblage de sous-unités potentiellement “libres”. Les isoformes de la sous-unité β se présentent souvent sous la forme de combinaisons de deux isoformes différentes dans un récepteur, ce qui introduit une plus grande incertitude quant à l’agencement précis des sous-unités dans les récepteurs natifs. Ceci est important pour le développement de médicaments car les interfaces des sous-unités abritent des sites de liaison sur lesquels des récepteurs spécifiques peuvent être ciblés de manière sélective, ou sur lesquels la liaison doit être évitée pour réduire les effets secondaires. On pense que les sous-unités ρ s’assemblent principalement entre elles (c’est-à-dire le groupe des récepteurs GABA-C) ; cependant, l’assemblage avec d’autres sous-unités a également été suggéré=. La composition et donc aussi la stœchiométrie et l’arrangement des récepteurs contenant ε ou θ restent inconnus. Le coassemblage de l’une ou l’autre des deux sous-unités avec α3 a été suggéré sur la base de la colocalisation dans des régions cérébrales spécifiques. La sous-unité π est exprimée principalement dans les cellules non neuronales, souvent avec β3, et on ne sait pas à quels assemblages pentamériques elle participe. |
| Encadré 3 Nomenclature. (i) Les récepteurs GABA-A α1β2γ2 (et tout autre récepteur contenant les sous-unités α1, 2, 3 ou 5) ont également été appelés “récepteurs α1”, “récepteurs α2”, etc. Étant donné que les principales formes de récepteurs GABA-A sont constituées de cinq sous-unités, avec souvent deux isoformes différentes de sous-unités α dans le pentamère, ces abréviations doivent être évitées ; selon la nomenclature recommandée par l’Union internationale de pharmacologie fondamentale et clinique (IUPHAR), toutes les sous-unités présentes dans un récepteur doivent être spécifiées. Seule la sous-unité α voisine de la sous-unité γ définit les propriétés du site de haute affinité pour les benzodiazépines. (ii) Les benzodiazépines classiques agissent comme des modulateurs allostériques positifs en se liant à un site de liaison à haute affinité présent dans certains types de récepteurs GABA-A. Ces composés ont également été appelés “agonistes” même s’ils n’ont que peu ou pas d’action agoniste sur les récepteurs GABA-A. Historiquement, le terme complet était “agoniste des récepteurs des benzodiazépines” et se référait uniquement aux effets “agonistes” sur le site de liaison des benzodiazépines. Si le site de liaison à haute affinité pour les benzodiazépines est occupé par un composé qui n’affecte pas la réponse au GABA, on parle d’antagoniste. Ce type de molécule est également appelé modulateur nul ou silencieux. Si le site est occupé par un composé qui entraîne une diminution de la réponse au GABA, on parle de modulateur allostérique négatif. Cette dernière catégorie de composés est également appelée “agonistes inverses”. |
Les récepteurs contenant une interface de sous-unité αx/γy, où x = 1, 2, 3, 5 et y = 1-3, forment un site de liaison à haute affinité pour les benzodiazépines (Figure 1A), pour les drogues Z développées ultérieurement de différents chimiotypes, et pour un grand nombre de composés de recherche chimiquement distincts (Encadré 1 pour des exemples). Pour les récepteurs contenant les sous-unités γ1 ou γ3, l’affinité et l’efficacité de nombreux ligands sont réduites. Les sous-unités α4 et α6 ne fournissent un site de liaison que pour une sélection limitée de composés, et sont donc traditionnellement appelées sous-unités insensibles au diazépam (ou DI) pour les distinguer des sous-unités sensibles au diazépam (DS). De même, de nombreuses isoformes de récepteurs GABA-A moins abondantes ne lient pas les benzodiazépines avec une grande affinité.
La majorité des récepteurs GABA-A sont exprimés dans les tissus neuronaux, mais on les trouve également dans de nombreux autres organes. Le potentiel thérapeutique des récepteurs dans le muscle lisse des voies aériennes pour soulager les spasmes de l’asthme bronchique est actuellement exploré. Cependant, nous nous concentrons ici sur les récepteurs neuronaux. Les récepteurs GABA-A situés au niveau de la synapse, dont on pense qu’ils sont principalement composés de récepteurs assemblés à partir de deux sous-unités α, deux sous-unités β et une sous-unité γ, médient des courants d’ions chlorure au niveau d’une synapse unique qui durent des millisecondes. Une autre classe de récepteurs, appelés récepteurs extrasynaptiques, est responsable de courants de chlorure qui durent des minutes et des heures dans de grandes parties du neurone entier, ajustant ainsi l’excitabilité de la cellule.
Bien que cette revue se concentre sur la modulation des récepteurs GABA-A par les ligands des sites de liaison des benzodiazépines, il convient de souligner que ces récepteurs répondent également à une grande variété d’autres modulateurs, y compris diverses toxines, les barbituriques, les anesthésiques intraveineux tels que le propofol et l’étomidate, les anesthésiques volatils tels que l’isoflurane, les neurostéroïdes, les insecticides et les composés végétaux.
Structure des récepteurs GABA-A.
Les récepteurs GABA-A appartiennent à la famille des récepteurs à boucle cys. Les structures cristallines de protéines homologues ont permis de mieux comprendre leur structure au niveau atomique. Tout d’abord, la protéine de liaison à l’acétylcholine de Lymnaea stagnalis a été analysée, suivie par des canaux ligand-gated pentamériques de bactéries et de C. elegans 28, 29, 30. Cependant, une structure à haute résolution du site de liaison des benzodiazépines dans le récepteur GABA-A fait toujours défaut. Un récepteur GABA-A β3 homomérique humain dépourvu de domaine intracellulaire a été cristallisé, mais il ne répond malheureusement ni aux benzodiazépines ni à l’agoniste naturel GABA. En raison de la grande homologie entre les sous-unités, il est néanmoins considéré comme une structure modèle réaliste pour l’architecture globale du site de liaison. Tous les pentamères de récepteurs à boucle cys contiennent de nombreuses cavités, et il a été démontré que les benzodiazépines interagissent avec de multiples sites distincts au niveau des localisations extracellulaires et transmembranaires (Figure 1B).
Classification des sites de liaison aux benzodiazépines.
Historiquement, les sites aux interfaces α+/γ- extracellulaires ont été désignés comme un seul “site de liaison aux benzodiazépines à haute affinité”, même si plusieurs isoformes de récepteurs GABA-A possèdent un tel site. Cela se justifie par le fait que de nombreux ligands se lient avec une puissance très similaire à la plupart ou à l’ensemble de ces isoformes de récepteurs. Ces sites de haute affinité sont formés par de multiples segments protéiques discontinus, appelés “boucles A-G” (figure 1C).
D’autres sites extracellulaires sont homologues au site canonique (figure 1B). L’interface α+/β- est structurellement similaire à l’interface α+/γ-. Il n’est donc pas surprenant que certains ligands du site benzodiazépine de haute affinité puissent également occuper cette interface homologue de la sous-unité. Ces composés (par exemple, CGS 8216 ; encadré 1) peuvent agir comme antagonistes à l’interface de la sous-unité α/γ et comme modulateurs allostériques positifs à l’interface de la sous-unité α/β, ou vice versa [34]. Un site supplémentaire de haute affinité pour le diazépam a récemment été localisé dans une position homologue à l’interface des sous-unités β2+/γ2-, ce qui n’est pas le cas des récepteurs αβγ.
En outre, des sites de faible affinité qui ne partagent aucune homologie structurelle avec les sites susmentionnés ont été décrits dans le domaine transmembranaire. On a découvert que, dans les récepteurs α1βγ2, la potentialisation des courants activés par le GABA par de fortes concentrations de diazépam est biphasique, avec une composante de haute et de basse affinité. La mutation combinée des résidus homologues α1S269, β2N265 et γ2S280 dans les seconds domaines transmembranaires (M2) en isoleucine a aboli cette composante micromolaire de la potentialisation alors que la composante de haute affinité n’a pas été affectée. Ces résidus font partie de cavités homologues à la cavité occupée par l’ivermectine dans les récepteurs GluCl cristallisés de C. elegans. Le récepteur GluCl homo-pentamérique héberge cinq sites ivermectine identiques, un à l’interface de chaque sous-unité. Dans les récepteurs GABA-A α1βγ2, il y a quatre interfaces différentes, chacune abritant la cavité correspondante (Figure 1B). Au moins trois interfaces différentes, voire toutes, des récepteurs α1βγ2 peuvent lier le diazépam. D’autres ligands de ce site ont été identifiés. Il est important de noter que la présence des sites non canoniques n’est pas limitée aux récepteurs αβγ, et que les récepteurs contenant la sous-unité δ sont également modulés par ces sites.
Comment les benzodiazépines agissent-elles au niveau moléculaire ?
Les ligands du site de haute affinité comprennent les modulateurs allostériques positifs, les modulateurs allostériques négatifs et les antagonistes (voir la nomenclature dans l’encadré 3). La présence de modulateurs allostériques positifs induit un déplacement de la courbe concentration-réponse du GABA vers des concentrations plus faibles. Inversement, les modulateurs allostériques négatifs la déplacent vers des concentrations plus élevées de GABA. Les modulateurs allostériques positifs déplacent l’équilibre entre l’état de repos lié au ligand et l’état pré-activé avant l’ouverture du canal, sans affecter l’amplitude maximale du courant déclenché par le GABA. Comme le montrent les études sur la liaison des agonistes aux ligands, ce phénomène s’accompagne d’une augmentation de l’affinité de l’agoniste. Les benzodiazépines affectent l’ouverture du canal des récepteurs GABA-A induite par l’un ou l’autre site de liaison de l’agoniste. Il n’existe pas d’analyse détaillée des ligands agissant sur les sites non canoniques. Au niveau macroscopique, ces ligands agissent également en tant que modulateurs allostériques positifs, modulateurs allostériques négatifs ou antagonistes.
Les “antagonistes” des sites benzodiazépines sont utilisés comme antidotes aux benzodiazépines et sont idéalement des modulateurs nuls, c’est-à-dire qu’ils se lient silencieusement sans augmenter ou réduire le transfert de charge déclenché par le GABA. Cependant, un modulateur nul parfait n’a pas encore été décrit. De nombreux composés qui agissent comme des antagonistes des benzodiazépines in vivo se sont révélés moduler au moins certaines isoformes de récepteurs dans des conditions expérimentales qui permettent d’étudier des récepteurs définis. Par exemple, le flumazénil, antidote des benzodiazépines utilisé en clinique, est un modulateur allostérique négatif partiel (faible). Son action dépend de la concentration. À de faibles concentrations, il agit comme un modulateur allostérique négatif faible, et à 1 μM, le flumazénil est un antagoniste pour les récepteurs exprimés dans les ovocytes de Xenopus. À des concentrations plus élevées, le flumazénil agit comme un modulateur allostérique positif faible.
L’occupation des sites allostériques peut favoriser l’ouverture, la désensibilisation ou la fermeture des récepteurs. Par conséquent, les ligands agissent comme des modulateurs allostériques positifs ou négatifs. Les récepteurs GABA-A existent dans au moins une conformation fermée, une conformation pré-activée liée à un ligand, une conformation ouverte et une conformation désensibilisée. Chacune de ces conformations prend d’autres formes différentes en présence de modulateurs allostériques positifs et négatifs ou d’antagonistes du site de liaison des benzodiazépines. Ainsi, les études cristallographiques ne permettent pas à elles seules de comprendre la complexité de la modulation allostérique, car les structures cristallographiques représentent des conformations statiques qui peuvent ne correspondre à aucune des conformations physiologiques.
Les sous-types de récepteurs et leur fonction – Évaluation critique.
Les ligands largement non sélectifs du site de liaison des benzodiazépines, tels que le diazépam, sont connus pour provoquer un large éventail d’effets in vivo, notamment l’hypnose, la sédation, l’anxiolyse et la relaxation musculaire. Des souris génétiquement modifiées ont été introduites dans le but d’attribuer des effets individuels à des récepteurs contenant des isoformes spécifiques de la sous-unité α. Il a été proposé que les effets sédatifs soient médiés spécifiquement par les “récepteurs α1” (encadré 3 pour la nomenclature préférée). Des tentatives ont été faites pour séparer les effets sédatifs des effets anxiolytiques, par exemple en réduisant ou en éliminant l’action sur les récepteurs α1 (“médicaments d’épargne α1”). Il a toutefois été souligné que les anxiolytiques non sédatifs ne sont généralement pas α1-sparing dans les essais fonctionnels utilisant des systèmes recombinants. D’autres lignes de recherche ont en fait corrélé les isoformes β avec l’efficacité des médicaments sédatifs, ataxiques ou narcotiques.
Les progrès technologiques récents permettent maintenant des approches orthogonales aux animaux transgéniques avec des sous-unités insensibles aux médicaments. Des manipulations optogénétiques ont été mises au point pour permettre le contrôle par la lumière de populations de récepteurs spécifiques, ce qui permettra de mieux cerner leurs rôles physiologiques. Toutefois, cette nouvelle approche visant à délimiter le rôle physiologique de sous-types de récepteurs définis nécessite des connaissances sur la composition et l’agencement des sous-unités de chaque espèce de récepteur, qui font encore largement défaut (encadré 2).
Étant donné qu’une grande partie des récepteurs GABA-A contiennent deux isoformes α ou β différentes dans un seul pentamère de récepteur, il serait surprenant qu’une seule sous-unité exprimée dans de nombreuses isoformes de récepteurs et dans différentes populations cellulaires puisse expliquer un seul effet comportemental d’un médicament. Pour illustrer cette complexité, nous examinons ici le cas des cellules granuleuses du cervelet (CGC). Comme indiqué ci-dessus, le site de liaison à haute affinité pour les benzodiazépines a été localisé à l’interface des sous-unités αx/γ où x = 1, 2, 3, 5, mais pas 4 ou 6. De nombreuses cellules expriment plusieurs isoformes de sous-unités. Les CGC expriment de nombreuses sous-unités, parmi lesquelles α1, α6, βx et γ2. Il a été démontré que les récepteurs α1α6βxγ2 dominent sur les récepteurs α6βxγ2. Les travaux sur les systèmes recombinants indiquent que les récepteurs α1γ2β2α1β2, α6γ2β2α6β2, α1γ2β2α6β2, et α6γ2β2α1β2 diffèrent tous dans leurs propriétés fonctionnelles et leur sensibilité aux médicaments. Seules les assemblées de récepteurs dont la sous-unité α1 est adjacente à la sous-unité γ2 répondent aux benzodiazépines. Comment la CCG, ou toute autre cellule, assemble-t-elle les récepteurs, et quels mécanismes conduisent à l’assemblage de pentamères spécifiques ? L’assemblage des récepteurs est-il dynamique ? La sensibilité aux médicaments peut-elle ainsi être modulée ? Toutes ces questions sont encore ouvertes à la recherche. Il a été récemment démontré que la modulation de la localisation et du recrutement des récepteurs dans les sites synaptiques et extrasynaptiques par les benzodiazépines se produit de manière bidirectionnelle. Cependant, le mécanisme de cette relocalisation reste à élucider.
Au-delà des efforts pour séparer les effets souhaitables établis (tels que l’anxiolyse) des effets concomitants (tels que la sédation dans le contexte de l’anxiolyse diurne) par un ciblage spécifique du sous-type, d’autres concepts thérapeutiques potentiels basés sur un ciblage sélectif du sous-type sont en cours d’étude. Il s’agit notamment des états douloureux, des troubles affectifs tels que la dépression et des déficits cognitifs dans la schizophrénie, les troubles du développement et les troubles neurodégénératifs. Dans le traitement de la dépression, la prescription simultanée d’antidépresseurs et de benzodiazépines a impliqué pendant un certain temps l’utilisation hors AMM de plusieurs benzodiazépines, mais l’efficacité des benzodiazépines dans le traitement de la dépression est probablement limitée à l’anxiolyse dans les cas où l’anxiété est parallèle à la dépression. Malheureusement, il y a une pénurie générale d’études cliniques qui permettraient une comparaison systématique des benzodiazépines individuelles dans les différentes indications neuropsychiatriques dans lesquelles elles sont largement utilisées hors AMM.
Des progrès récents ont été réalisés dans la traduction des découvertes animales sur les performances de la mémoire et les effets amnésiques – qui sont largement déterminés par des récepteurs toniquement actifs dans lesquels α5 contribue au site de liaison de la benzodiazépine. Sur la base de l’observation que la suppression de cette sous-unité conduisait à une amélioration des performances d’apprentissage spatial, l’hypothèse a été émise que les effets amnésiques des benzodiazépines (également observés dans les anesthésiques sédatifs) pouvaient être attribués à une activité excessive de cette population de récepteurs, et que les effets nootropes pouvaient être provoqués par des modulateurs allostériques négatifs sélectifs de l’α5. Cette notion a été confirmée par des études animales utilisant plusieurs composés expérimentaux. Cette ligne de recherche a donné naissance au basmisanil (RG1662), le modulateur négatif α5-sélectif le plus avancé à ce jour. Ce composé a été testé dans le cadre d’un essai clinique sur une cohorte de personnes atteintes du syndrome de Down, afin de déterminer s’il pouvait atténuer les troubles cognitifs. Aucun bénéfice n’a été constaté, mais le composé peut améliorer la cognition chez les sujets sains.
Les déficits cognitifs sont également des symptômes négatifs chez les patients schizophrènes, et les benzodiazépines sont depuis longtemps des candidats pour cette indication également. Un essai à petite échelle avec le bretazénil a été interrompu en raison d’effets sédatifs excessifs malgré une efficacité prometteuse. Les récepteurs contenant les sous-unités α2 et α5 étaient traditionnellement considérés comme les cibles les plus prometteuses pour traiter les symptômes de la schizophrénie. Au moment de la rédaction de ce document, le basmisanil est testé dans une cohorte de patients schizophrènes en complément d’un traitement antipsychotique afin de vérifier si les symptômes négatifs peuvent être réduits.
Dans l’ensemble, le ciblage sélectif des sous-types a été développé avec succès dans des modèles de rongeurs. La preuve de concept chez l’homme est également bien établie pour certaines espèces de récepteurs – comme les récepteurs contenant la sous-unité α5 en tant que cibles pour les effets nootropes ou amnésiques. La transposition des résultats obtenus chez l’animal à l’homme reste un défi, et les échecs des essais peuvent résulter de différences de séquence substantielles dans plusieurs sous-unités de récepteurs qui ont un impact sur les effets des médicaments, ainsi que de différences dans les schémas d’expression des sous-unités individuelles dans des environnements anatomiques et cellulaires distincts. En outre, la composition et la disposition des sous-types de récepteurs individuels peuvent également ne pas être identiques dans les différentes espèces pour tous les sous-types de récepteurs.
Nouveaux ligands pour les sites de liaison à haute affinité des benzodiazépines.
Le développement des benzodiazépines était traditionnellement basé sur de grandes séries de ligands et sur l’exploration expérimentale des relations structure-activité (quantitatives). Il y a encore peu de composés sélectifs, et cette lacune pourrait être comblée par la conception de ligands guidée par la structure. L’encadré 4 passe en revue les principes de la conception de médicaments guidée par la structure. Les modèles informatiques basés sur les protéines homologues disponibles sont globalement en accord avec les positions relatives des segments protéiques individuels dans le site de liaison. Sur la base des premiers travaux de mutation, ces segments ont été initialement appelés “boucles” A-G, certains étant des boucles au sens structurel, tandis que d’autres sont de courts morceaux de brins β. L’utilisation de tels modèles d’homologie, en combinaison avec une approche de biologie chimique pour identifier les résidus en contact avec le diazépam, a permis le positionnement relatif du diazépam dans les structures d’homologie d’un récepteur GABA-A α1γ2β2α1β2. Le criblage virtuel de ligands dans cette structure a conduit à la découverte de nouveaux ligands ayant une grande affinité pour le site de liaison canonique des benzodiazépines, validant ainsi l’exactitude globale des modèles structurels.
| Encadré 4 Principes de la conception de médicaments guidée par la structure La conception de médicaments guidée par la structure est possible grâce à des modèles structurels fiables des poches de liaison. Sur la base d’un modèle atomique du site de liaison, des structures multiples permettent d’explorer des facteurs tels que les régions localement variables et les différentes conformations de la poche. Si ces structures ne sont pas accessibles, des approximations peuvent être obtenues à partir de prédictions informatiques. Pour les sites de benzodiazépines, le processus est actuellement limité aux modèles d’homologie. L’une des limites est que la structure connue de la protéine impose une similarité excessive à la structure du modèle. Ce sera le cas si une sous-unité α est modélisée à partir de la structure disponible de la sous-unité β. En principe, la poche sans ligand peut être exploitée. Cependant, une poche ligaturée permet de s’assurer que le ligand peut être logé stériquement et que des interactions favorables stabilisent l’état lié. D’autre part, les poses de ligands (d’origine expérimentale ou informatique) nécessitent une validation supplémentaire et ne reflètent pas nécessairement la structure pharmacologiquement active. Des preuves expérimentales indirectes peuvent fournir des informations sur l’orientation du ligand dans la poche, comme les réactions covalentes accélérées par la proximité entre les ligands fonctionnalisés et les poches fonctionnalisées. Cela permet de valider les modèles d’état lié. Le criblage in silico de composés candidats dans des représentations d’états liés peut être réalisé par différents moyens. Dans notre cas, une approche basée sur le pharmacophore utilisant le programme LigandScout s’est avérée efficace pour fournir de nouveaux échafaudages de haute affinité. Le complexe ligand-poche est représenté par un modèle pharmacophore abstrait. Les propriétés stériques de la poche peuvent être représentées de manière à ce que les parties moins fiables ou flexibles de la poche n’imposent pas de contraintes excessives sur la correspondance des molécules avec le pharmacophore. Les interactions entre le(s) ligand(s) de référence et la poche sont décrites comme des “caractéristiques pharmacophoriques” abstraites avec des positions définies et peuvent inclure des groupements hydrophobes, des interactions aromatiques, des interactions cation-π, des caractéristiques acceptant et donnant des liaisons hydrogène et des interactions chargées (ponts salins). Il est bon d’utiliser des modèles de pharmacophores dérivés de plusieurs liants connus pour tenir compte de l’utilisation alternative des valences d’interaction dans la poche. Certains algorithmes permettent également d’inclure des molécules d’eau médiatrices de liaisons hydrogène qui peuvent être des molécules d’eau essentielles dans les états liés. Bien que les algorithmes de découverte de médicaments in silico basés sur la structure génèrent, en plus des vrais résultats, une série de résultats faussement positifs et faussement négatifs, ils permettent néanmoins de réaliser des économies significatives en limitant le criblage expérimental aux résultats in silico au lieu de nécessiter de grandes bibliothèques. En outre, les structures modèles peuvent être testées expérimentalement (par exemple, analyse mutationnelle). Les avantages évidents sont la présélection de molécules pour des tests expérimentaux en aval, et l’hypothèse structurelle testable pour des travaux ultérieurs. |
En raison de la grande conservation des boucles A-G dans les sous-unités individuelles, telles que α2 et α3, ou γ2 et γ3, l’architecture globale des sites individuels est similaire dans les différentes isoformes du récepteur. Seuls quelques acides aminés dans le site de liaison confèrent des propriétés uniques à la poche pour permettre la sélectivité du ligand (Figure 1C,D). En raison de limitations méthodologiques, les modèles de protéines hautement homologues (telles que α2 et α3) basés sur un membre de la famille plus éloigné (tel que β3) ne prédiront pas de manière fiable les différences subtiles entre les isoformes individuelles des sites de liaison à haute affinité pour les benzodiazépines.
Les sites de liaison canoniques auxquels contribuent α2, α3 ou α5 sont très similaires. Par conséquent, les différences d’affinité du ligand ne seront pas importantes même si un ligand utilise de manière optimale les petites différences entre les poches. Une autre approche possible pour séparer les effets des composés consisterait à utiliser des ligands ayant une affinité de liaison similaire mais des effets allostériques différents sur différents sous-types de récepteurs (ligands fonctionnellement sélectifs). Idéalement, un ligand fonctionnellement sélectif devrait se lier silencieusement (comme un antagoniste) à tous les sous-types sauf un, où il devrait exercer des effets modulateurs positifs ou négatifs. Bien que ce concept ait donné lieu à des résultats prometteurs, il reste à déterminer si la liaison “silencieuse” est effectivement physiologiquement silencieuse et n’entraînera pas de changements adaptatifs indésirables à long terme dans le système nerveux. En outre, le développement de tels ligands guidé par la structure est rendu difficile pour deux raisons : (i) les essais fonctionnels prennent beaucoup plus de temps que les essais de liaison, et (ii) les structures du site de liaison des benzodiazépines dans les états apo et positivement allostériques stimulés ne sont pas connues.
Les sites de liaison non canoniques peuvent constituer des cibles intéressantes.
Comme indiqué ci-dessus, outre le site de haute affinité pour les benzodiazépines, plusieurs sites non canoniques sont présents aux interfaces des sous-unités et dans la partie transmembranaire du récepteur. En raison de la grande similitude, par exemple entre γ2 et γ3, ou entre α2 et α3, leurs sites de liaison à haute affinité pour les benzodiazépines ne conviennent pas pour un ciblage sélectif fiable, ni pour une sélectivité de liaison ou fonctionnelle. L’utilisation d’autres sites de liaison allostérique pourrait être une alternative intéressante. Pour les sites de liaison α+/β- extracellulaires, des ligands β1-sélectifs d’une puissance de l’ordre du nM ont déjà été rapportés.
Les sites des domaines transmembranaires conviennent généralement moins bien aux ligands sélectifs de sous-types en raison de la plus grande conservation de la séquence et de la structure dans cette partie des sous-unités. Cependant, certaines sous-unités spécifiques présentent des segments de poche uniques dans ce groupe de sites de liaison allostérique. Par exemple, les sous-unités α2 et α3, qui ont des côtés extracellulaires plus presque identiques (conduisant ainsi à une pharmacologie des benzodiazépines de haute affinité très similaire), contiennent des segments transmembranaires M3 et M4 distincts qui peuvent potentiellement être ciblés individuellement par des ligands appropriés.
Jusqu’à présent, les données limitées (qui ne concernent que le diazépam) rendent difficile la prédiction détaillée de l’action des médicaments par l’intermédiaire des sites non canoniques. Des souris porteuses d’une mutation ponctuelle qui rend le site de liaison canonique insensible au diazépam dans les quatre sous-unités α du DS ont été étudiées. Ces souris étaient protégées contre la relaxation musculaire et la déficience motrice induites par le diazépam. Ces souris ont montré, sous traitement, une activité locomotrice réduite qui était relativement importante à des doses plus élevées. Cela indique qu’au moins une partie de cette réponse résiduelle au diazépam pourrait être médiée par des sites non canoniques. Il convient de noter que des effets mineurs peuvent facilement passer inaperçus dans les expériences comportementales.
Remarques finales.
Les recherches futures porteront sur plusieurs domaines majeurs. Tout d’abord, la composition et la fonction exactes des isoformes des récepteurs doivent être étudiées plus en détail. Alors que les effets biologiques des récepteurs contenant différentes isoformes de sous-unités α sont progressivement élucidés, on ne sait toujours pas dans quelles espèces de récepteurs (sous-types) ils sont assemblés, ni quels sont les rôles des isoformes γ individuelles en termes de physiologie et de pharmacologie des récepteurs. On ne sait pas non plus si des espèces de récepteurs homologues exercent des fonctions physiologiques homologues chez l’homme et chez l’animal de laboratoire. Pour répondre à certaines de ces questions, il est nécessaire de déterminer la composition exacte des récepteurs et la disposition des sous-unités pour toutes les isoformes de récepteurs qui médient les effets des benzodiazépines (voir Questions en suspens).
Deuxièmement, une recherche clinique plus poussée permettrait de mieux comprendre et de quantifier les différences entre des médicaments pharmacologiquement similaires, et fournirait des preuves scientifiques aux “pratiques de prescription” qui diffèrent souvent de façon marquée entre les pays et même entre les institutions d’un même pays. En outre, la recherche clinique systématique permettrait de mieux définir les besoins médicaux les plus urgents auxquels il conviendrait de répondre dans le cadre de la recherche fondamentale et translationnelle. L’établissement des préférences de sous-type des benzodiazépines utilisées en clinique sur des récepteurs définis, exprimés par recombinaison et contenant des sites de liaison canoniques et non canoniques, pourrait aider à combler le fossé entre les résultats précliniques et cliniques, et permettrait de mettre en correspondance les sites de liaison des médicaments avec les différents effets souhaités et non souhaités des benzodiazépines.
Troisièmement, la conception rationnelle de médicaments avec des profils de sous-types mieux définis est un besoin urgent. Alors qu’un nombre énorme de petites molécules de ∼100 chimiotypes ont été identifiées comme ligands des sites de liaison canoniques des benzodiazépines, pour la majorité d’entre elles, les profils de sous-types ne sont que partiellement connus. À l’heure du haut débit et de la science des données massives, il devrait être possible de mettre en place un référentiel structuré et des protocoles normalisés pour la détermination de la caractérisation électrophysiologique complète des effets des composés sur un large panel d’isoformes de récepteurs et sur leurs sites de liaison respectifs aux benzodiazépines. Ces données permettraient alors d’identifier les composés les plus prometteurs en vue d’une optimisation ultérieure afin d’obtenir des ligands présentant des préférences nouvelles ou améliorées pour les sous-types. Les méthodes in silico de découverte de médicaments, telles que le criblage de pharmacophores basé sur la structure des bibliothèques, complétées par des données expérimentales, permettraient d’accélérer encore le développement de composés de base en outils de recherche utiles, voire en nouveaux médicaments thérapeutiques.
Quatrièmement, les sites non canoniques peuvent être ciblés. Les benzodiazépines classiques nécessitent la présence d’une sous-unité γ pour une liaison de haute affinité, ce qui limite leur activité à un large groupe spécifique d’isoformes de récepteurs, les autres isoformes n’étant pas affectées. En particulier, les récepteurs contenant une sous-unité δ, ainsi que des populations de récepteurs moins étudiées telles que les récepteurs contenant une sous-unité θ ou ε, sont considérés comme médiateurs de fonctions physiologiques très spécifiques et pourraient donc être des cibles potentiellement intéressantes pour de nouvelles approches thérapeutiques. Les sites benzodiazépines non canoniques sont présents sur une large gamme de récepteurs car ils peuvent également être formés par les sous-unités α et β. Étant donné que l’échafaudage des benzodiazépines présente un profil toxicologique extrêmement bénin et d’excellentes propriétés ADMET (absorption, distribution, métabolisme, excrétion et toxicité), le développement d’un certain degré de spécificité pour l’activité des sites non canoniques offre une voie très précieuse pour explorer une nouvelle chimie médicinale sur la base de médicaments déjà établis.
On peut donc s’attendre à ce que le récepteur GABA-A et ses sites de liaison aux médicaments jouent un rôle de premier plan dans la recherche de traitements pour les maladies du système nerveux central.
