
Kuropka, P., Zawadzki, M., & Szpot, P. (2023). A narrative review of the neuropharmacology of synthetic cathinones—Popular alternatives to classical drugs of abuse. Human Psychopharmacology: Clinical and Experimental, e2866.
Abstract
Objectif de l’étude
Examiner la littérature sur la neuropharmacologie des cathinones synthétiques.
Méthodes
Une recherche documentaire complète a été effectuée dans plusieurs bases de données (principalement PubMed, World Wide Web et Google Scholar) à l’aide de mots-clés pertinents.
Résultats
Les cathinones présentent un large profil toxicologique, imitant les effets d’une grande variété de “drogues classiques” telles que la 3,4-méthylènedioxyméthamphétamine (MDMA), la méthamphétamine et la cocaïne. Même de petites modifications structurelles affectent leurs interactions avec des protéines clés. Cet article passe en revue les connaissances actuelles sur les mécanismes d’action des cathinones au niveau moléculaire, ainsi que les principaux résultats de la recherche sur leur relation structure-activité. Les cathinones sont également classées en fonction de leur structure chimique et de leur profil neuropharmacologique.
Conclusions
Les cathinones synthétiques représentent l’un des groupes les plus nombreux et les plus répandus parmi les nouvelles substances psychoactives. Initialement développées à des fins thérapeutiques, elles ont rapidement commencé à être utilisées à des fins récréatives. Avec l’augmentation rapide du nombre de nouveaux agents arrivant sur le marché, les études sur les relations structure-activité sont précieuses pour évaluer et prédire le potentiel de dépendance et la toxicité des substances nouvelles et potentielles. Les propriétés neuropharmacologiques des cathinones synthétiques ne sont pas encore totalement comprises. L’élucidation complète du rôle de certaines protéines clés, y compris les transporteurs de cations organiques, nécessite des études détaillées.
1. INTRODUCTION
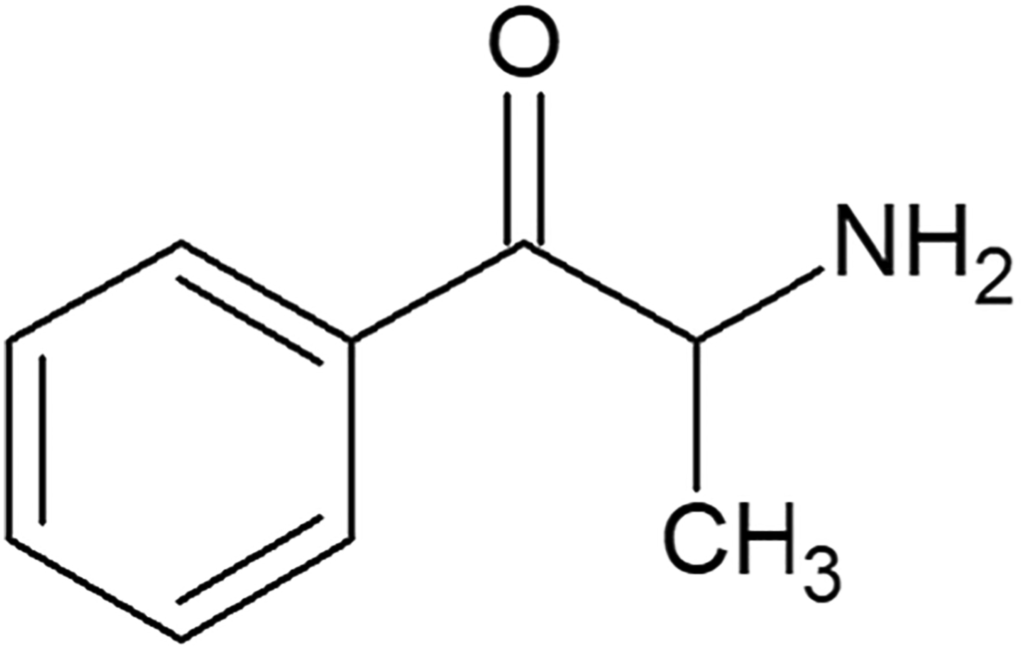
La cathinone (2-amino-1-phénylpropan-1-one) est un alcaloïde naturellement présent dans le khat (Catha edulis) (figure 1). En tant que β-cétone analogue de l’amphétamine, la cathinone est appelée “amphétamine naturelle” en raison de sa structure similaire et de son effet stimulant. L’éphédrone (2-[méthylamino]-1-phénylpropan-1-one), un dérivé N-méthyle de la cathinone synthétisé pour la première fois en 1928 par oxydation de l’éphédrine (2-[méthylamino]-1-phénylpropan-1-ol), est considérée comme la première cathinone synthétique (CS). À l’heure actuelle, les CS constituent l’un des groupes les plus importants parmi les nouvelles substances psychoactives (NPS), qui sont conçues pour ressembler aux “drogues classiques” et leur offrir une alternative moins coûteuse. Le rapport de l’Observatoire européen des drogues et des toxicomanies (OEDT) indique que fin 2021, sur les 880 NPS surveillées, pas moins de 162 étaient des CS, tandis qu’en 2020 en Europe, environ deux tiers du matériel NPS saisi (plus de 3 tonnes) étaient des dérivés de la cathinone. Parallèlement, à l’échelle mondiale, un total de 201 CS ont été signalés au système d’alerte précoce de l’Office des Nations unies contre la drogue et le crime à la fin de l’année 2021. La plupart d’entre eux sont produits en Chine et dans d’autres pays asiatiques et arrivent sur le marché sans aucun test toxicologique ou pharmacologique. Un tel produit peut contenir de nombreuses substances actives du même groupe ou être un mélange de composés de nature chimique différente, ce qui peut entraîner un empoisonnement involontaire. La littérature fait état de nombreux cas d’intoxications graves, voire mortelles, par des CS. Le nombre de nouveaux dérivés disponibles sur le marché peut continuer à augmenter, c’est pourquoi l’étude des propriétés de chaque structure individuelle et l’obtention de son profil toxicologique complet est un processus laborieux, chronophage et inefficace. Des recherches sont en cours pour permettre de prédire les profils pharmacologiques des dérivés nouvellement développés et de ceux qui pourraient l’être à l’avenir. Cet article passe en revue la structure chimique des CS et les caractéristiques de leur action neuropharmacologique. Il résumera également les connaissances actuelles sur l’activité des CS en fonction de leur structure chimique.
2. STRUCTURE CHIMIQUE DES CATHINONES SYNTHÉTIQUES
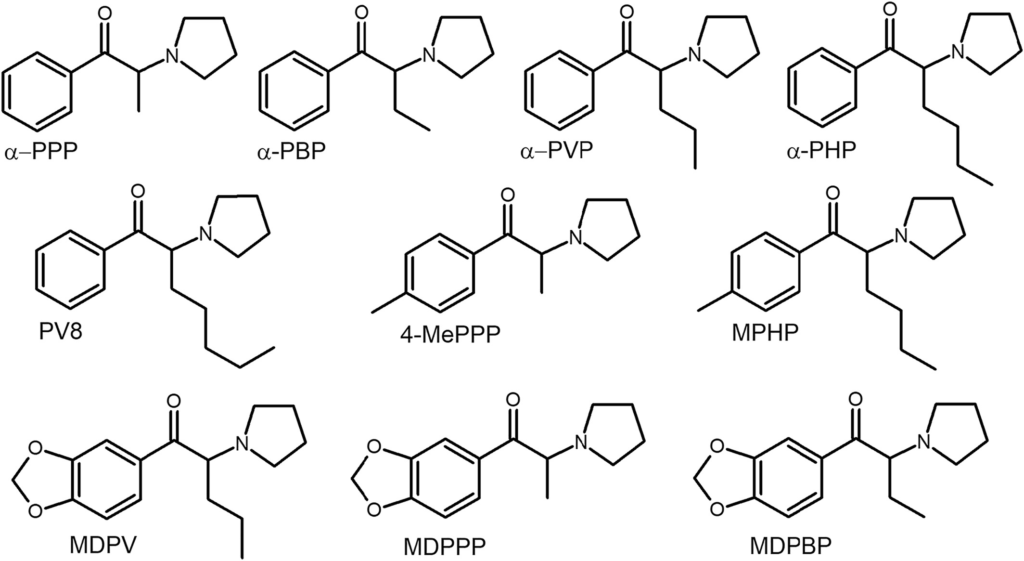
D’un point de vue chimique, tous les CS sont des dérivés de la cathinone. Ils ressemblent aux dérivés de l’amphétamine, sauf qu’il y a un groupe carbonyle en position β de la chaîne aminoalkyle. Tous les dérivés possibles sont formés en attachant des substituants à la structure de base de la cathinone à plusieurs positions typiques (figure 2).
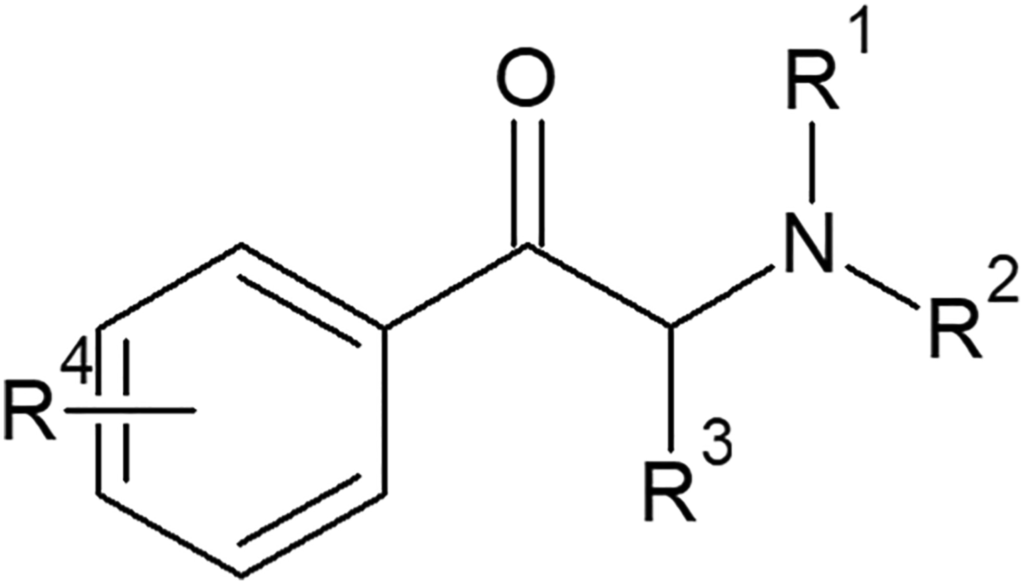
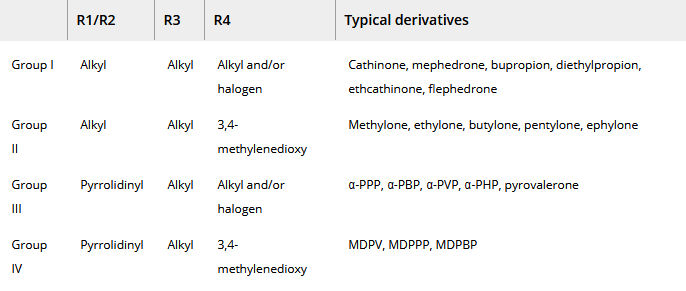
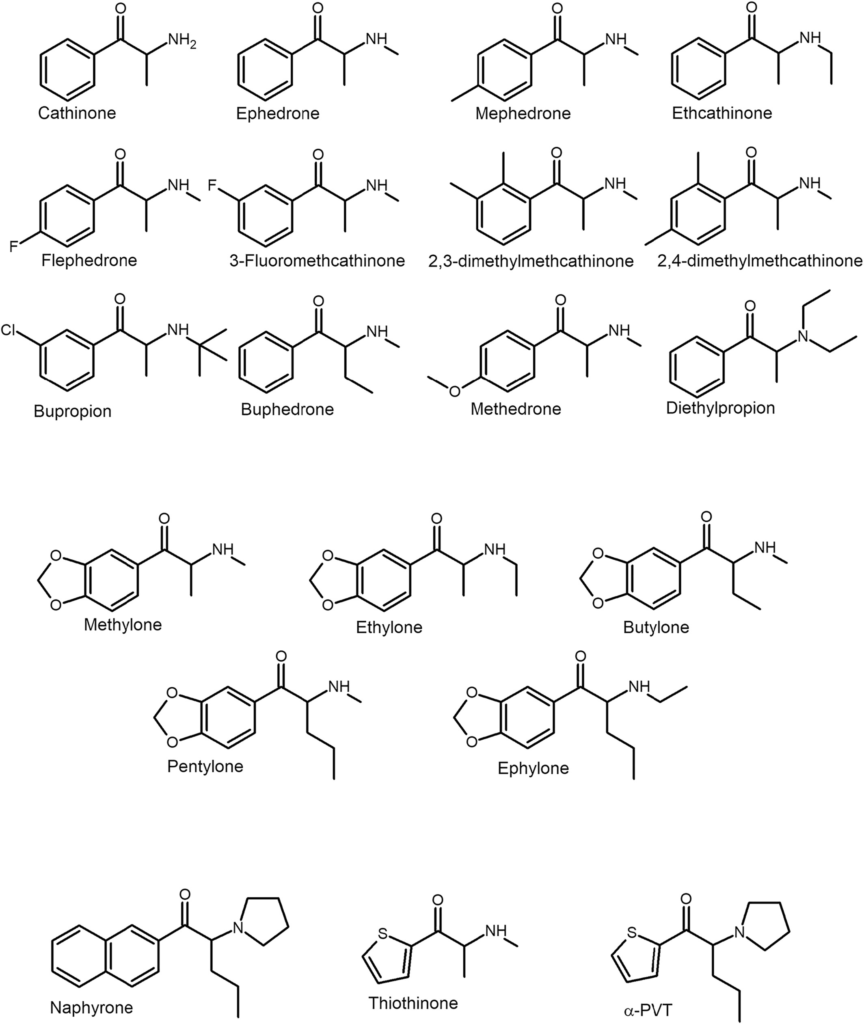
D’un point de vue structurel, les dérivés de la cathinone peuvent être classés en quatre groupes (tableau 1). La plupart de ceux qui ont été synthétisés initialement (y compris ceux qui ont des propriétés thérapeutiques – bupropion [2-{tert-butylamino}-1-{3-chlorophényl}propan-1-one] et diéthylpropion [2-diéthylamino-1-phénylpropan-1-one]) peuvent être classés dans le groupe le plus simple, le groupe I. Il s’agit de dérivés N-alkylés en position R1 et/ou R2. Ce groupe peut être substitué par un groupe alkyle (généralement méthyle ou éthyle) ou halogène en position R4 (figure 3). Les CS du groupe II ont une structure similaire à celle de la 3,4-méthylènedioxyméthamphétamine (MDMA), un dérivé populaire de l’amphétamine. Ils se distinguent par un substituant 3,4-méthylènedioxy en position R4 (figure 3). Les CS du groupe III ont un cycle pyrrolidine à la place du groupe amino et, comme pour le premier groupe, leur cycle aromatique est parfois substitué par des groupes alkyle ou halogène (figure 4). Le dernier groupe est une combinaison des groupes II et III. Les CS de ce groupe se caractérisent à la fois par la substitution du cycle aromatique par un groupe 3,4-méthylènedioxy et par la possession d’un cycle pyrrolidine. Les groupes III et IV sont souvent appelés conjointement “dérivés de pyrovalérone” ou “pyrovalérones”. Les dérivés de tous les groupes ont des longueurs de chaîne alkyle différentes en position R3 (Figure 4). Il existe également plusieurs dérivés qui ne peuvent être classés dans aucun des groupes. Il s’agit de substances formées en remplaçant un groupe phényle par un autre cycle (thiophène ou naphtyle) (figure 3). Il convient de noter que l’atome de carbone auquel est attaché le groupe amino constitue le centre stéréogène de la molécule, mais la grande majorité des produits CS existent sous forme racémique.
3. MÉCANISME D’ACTION DES CATHINONES SYNTHÉTIQUES
La prise d’une cathinone affecte la neurotransmission cérébrale dépendante des monoamines (dopamine [DA], noradrénaline [NE] et sérotonine [5-HT]), entraînant un effet psychostimulant similaire à celui de l’amphétamine. Cet effet est principalement dû à la capacité des stimulants à interagir avec des protéines membranaires présentes dans les neurones, appelées transporteurs de monoamines (MAT). Il existe trois principaux types de MAT dans le système nerveux central : le transporteur de dopamine (DAT), le transporteur de noradrénaline (NET) et le transporteur de sérotonine (SERT). Le rôle de ces protéines spécialisées est de recapter les neurotransmetteurs de la fente synaptique (libérés lors de la transmission du signal) dans le neurone. Dans des conditions physiologiques, cette recapture est le principal mécanisme d’inactivation de la signalisation. Les transporteurs vésiculaires de monoamines (VMAT) constituent un autre groupe important de protéines impliquées dans le transport des monoamines à l’intérieur du neurone. Les VMAT sont des protéines membranaires des vésicules synaptiques et sont responsables de la recapture des monoamines du cytoplasme dans la vésicule. L’interaction entre les MAT et les VMAT détermine la bonne circulation et la réutilisation des neurotransmetteurs (figure 5). De nombreux stimulants agissent en interagissant avec les protéines MAT et VMAT.
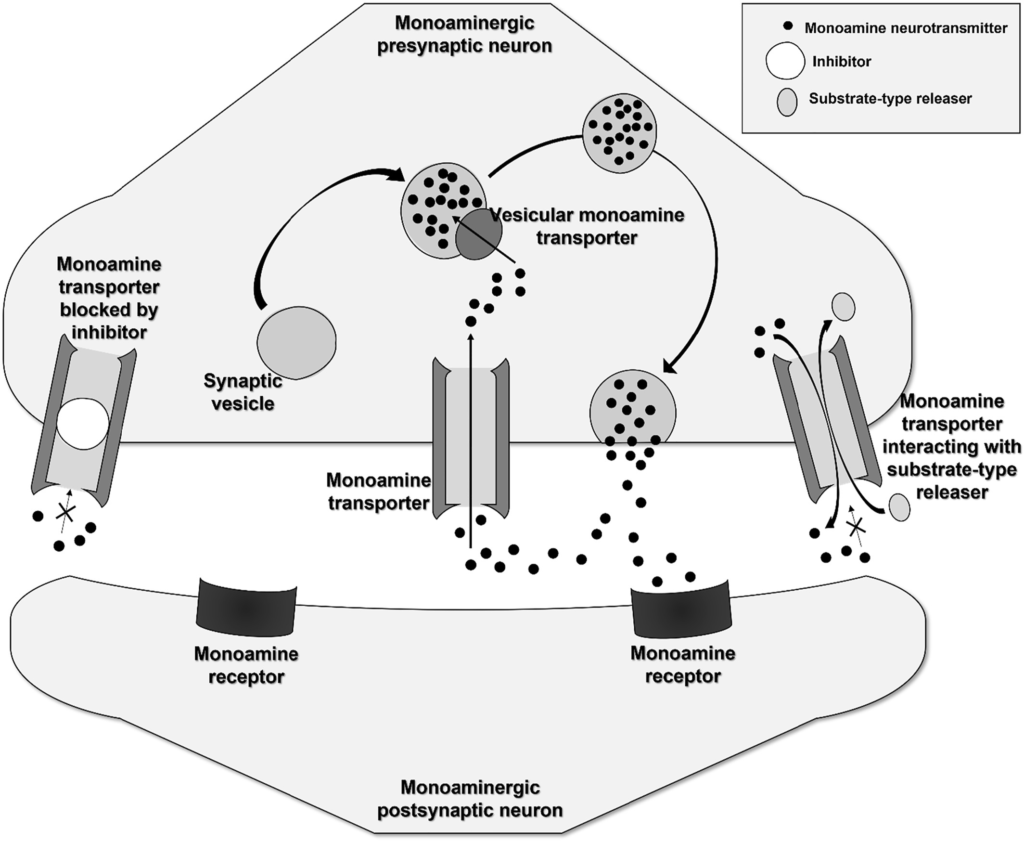
L’interaction des CS avec les protéines de la MAT peut varier. Même de petites modifications structurelles ont un impact important à la fois sur la puissance et sur la sélectivité de la protéine et, par conséquent, les CS individuels présentent des effets pharmacologiques différents, nécessitent des doses différentes pour induire une réponse psychoactive et présentent un potentiel addictif différent. Les CS peuvent interagir avec les MAT de deux manières différentes. Certains CS agissent comme des inhibiteurs (bloqueurs) en se fixant aux MATs et en les bloquant. Une augmentation de la concentration de neurotransmetteurs dans la fente synaptique est due au blocage de la recapture. D’autres CS agissent comme des libérateurs de type substrat pour les MAT. Ces substrats sont capables de passer à travers les MATs dans la cellule. En plus de l’inhibition compétitive de la recapture, les substrats inversent la direction standard du flux de neurotransmetteur. Ceci peut être facilité par l’interaction avec la protéine VMAT. Grâce à cette interaction, l’accumulation des neurotransmetteurs dans les vésicules synaptiques est entravée et, par la suite, les monoamines en excès présentes dans le cytosol sont libérées de la cellule par les MAT. Cela crée un mécanisme de libération des monoamines dans la fente synaptique qui diffère de la libération vésiculaire. Qu’un CS agisse comme un libérateur ou un bloqueur de type substrat, l’effet est similaire – une augmentation de la concentration extracellulaire de monoamine et donc une stimulation accrue du neurone postsynaptique (Figure 5).
Les effets pharmacologiques de la plupart des CS sont principalement dus à une augmentation des concentrations extracellulaires de monoamines endogènes. Par rapport aux amphétamines, les CS sont moins susceptibles d’interagir avec les récepteurs des monoamines. Par exemple, les hallucinogènes dérivés des amphétamines interagissent souvent directement avec les récepteurs de la sérotonine en les activant. Dans le cas des CS aux propriétés sérotoninergiques, leurs effets ne sont pas dus à une activation directe des récepteurs, mais plutôt à une action indirecte en augmentant la 5-HT endogène dans l’espace extracellulaire. Certains d’entre eux (en particulier les para-substitués) ont la capacité de se lier aux récepteurs de la sérotonine, mais ils n’agissent généralement pas comme leurs agonistes ou antagonistes fonctionnels et leur influence sur l’effet pharmacologique final est négligeable en raison de leurs faibles affinités de liaison. Il existe toutefois des dérivés (notamment la 2,3-diméthylméthcathinone [1-{2,3-diméthylphényl}-2-{méthylamino}propan-1-one] et la méphédrone [2-méthylamino-1-{4-méthylphényl}propan-1-one]) qui, lors d’un test d’activité in vitro (essai de mobilisation du calcium), ont montré une activation significative des récepteurs hallucinogènes 5-HT2A équivalente ou supérieure à celle de la MDMA). Ces résultats sont cohérents avec les observations de propriétés hallucinogènes induites chez les consommateurs de méphédrone. En revanche, pour d’autres dérivés (α-PPP [1-phényl-2-{1-pyrrolidinyl}-1-propanone] et 4-MePPP [1-{4-méthylphényl}-2-{1-pyrrolidinyl}-1-propanone]), une liaison à la micromole et une activité antagoniste contre le récepteur 5-HT2A humain ont été mises en évidence par un test de liaison par compétition de radioligands in vitro et un test au monophosphate d’inositol (IP-One), et cette activité pour l’α-PPP a été corroborée par des études in vivo (réduction de la réaction de contraction de la tête chez les souris induite par l’agoniste du récepteur 5-HT2A) (Chen et al. , 2019). Dans une autre étude impliquant l’α-PPP et le 4-MePPP, ainsi que deux autres dérivés MPHP (1-[4-méthylphényl]-2-[1-pyrrolidinyl]-1-hexanone) et MDPPP (1-[3,4-méthylènedioxyphényl]-2-[1-pyrrolidinyl]-1-propanone), une liaison micromoléculaire significative a été observée avec l’α-PPP et le 4-MePPP, Une liaison significative à la micromole au récepteur 5-HT2A humain a été confirmée par l’essai de liaison du radioligand, et il a été démontré par l’essai au calcium que, malgré la liaison, ces CS n’ont pas réussi à activer ces récepteurs. En outre, certains CS tels montrent une faible affinité pour les récepteurs α1A- et α2A-adrénergiques, qui sont responsables des effets sympathomimétiques. D’autre part, l’α-PHP (1-phényl-2-(pyrrolidine-1-yl)-hexan-1-one) dans les études in vitro interagit de manière antagoniste avec une autre cible en bloquant les récepteurs muscariniques humains M1 et M2 et a donc le potentiel de provoquer des signes et des symptômes anticholinergiques. Il est intéressant de noter qu’à ce jour, aucune étude in vitro n’a montré d’affinité pour les récepteurs D1, D2 ou D3 pour l’un des CS, de sorte que leurs effets dopaminergiques découlent uniquement de leur interaction avec le DAT.
Une autre caractéristique qui distingue les CS des amphétamines est leur interaction négligeable avec le récepteur 1 associé aux amines traces (TAAR1). L’activation de ce récepteur réduit l’activité des neurones dopaminergiques, réduisant ainsi les effets psychostimulants et le potentiel addictif. Les amphétamines sont de puissants agonistes de ce récepteur, ce qui les rend susceptibles d’auto-inhiber leurs effets stimulants. En revanche, les CS présentent une activité négligeable vis-à-vis de TAAR1. Les deux exceptions sont la 2,4-diméthylméthcathinone (1-[2,4-diméthylphényl]-2-[méthylamino]propan-1-one) et la 2,3-diméthylméthcathinone, qui ont montré une grande affinité pour les récepteurs TAAR1 du rat et de la souris lors d’un essai de liaison par radioligand. Il convient toutefois de noter que pour TAAR1, il existe une variabilité considérable entre les espèces dans son interaction avec les ligands, et il est possible que l’activité in vitro de ces CS ne se traduise pas par une activité dans le corps humain. L’absence d’autorégulation par TAAR1 pourrait en partie expliquer le potentiel addictif plus élevé des CS par rapport aux amphétamines.
Une question neurochimique intéressante est l’interaction entre les composés agissant comme des bloqueurs et des substrats. Les substrats (par exemple la méphédrone) utilisent les protéines des MAT pour libérer des neurotransmetteurs, tandis que les inhibiteurs (par exemple le MDPV [1-{1,3-benzodioxol-5-yl}-2-{pyrrolidin-1-yl}pentan-1-one]) empêchent le transport de tout composé à travers les MAT. Il s’ensuit que l’utilisation simultanée d’un substrat et d’un bloqueur devrait entraîner un mécanisme antagoniste qui réduit la puissance mutuelle. Paradoxalement, le MDPV et la méphédrone sont présents ensemble dans les mélanges et, en outre, ils renforcent leurs effets respectifs, ce qui entraîne une très forte stimulation. L’utilisation simultanée des deux CS est rapportée par les usagers, et des études chez les rongeurs impliquant l’administration simultanée des drogues ont montré une augmentation significative de l’activité locomotrice et un effet additif par rapport à l’administration de ces drogues seules. Initialement, sur la base d’études in vitro portant sur les courants électriques conduits par les DAT exprimés dans les ovocytes de Xenopus laevis, l’action synergique de ces deux CS a été expliquée comme suit : d’abord, la méphédrone aurait induit un transport inverse des monoamines à travers leurs transporteurs, et ce n’est qu’ensuite que le MDPV aurait empêché la recapture des monoamines libérées en bloquant les MAT. Cependant, même à de très faibles concentrations, le MDPV présente une affinité si forte pour les MAT qu’il est peu probable que la méphédrone puisse inverser le transport des monoamines en sa présence ; en outre, les observations de l’activité locomotrice chez le rat n’indiquent qu’une interaction additive de ces CS. L’explication de ce phénomène se trouve dans l’action des protéines transporteuses de cations organiques (OCT). Les OCT sont une famille de protéines responsables du transport endothélial de petites molécules organiques, hydrophiles et chargées positivement, dont les neurotransmetteurs et les xénobiotiques. OCT3 est une protéine présente dans les régions dopaminergiques du système nerveux central, où elle favorise la recapture de la DA lorsqu’elle est inhibée pour les transporteurs à haute affinité (DAT). Les monoamines peuvent également être libérées par les OCT. Ce transport bidirectionnel signifie que les OCT peuvent jouer un rôle important dans les mécanismes des xénobiotiques qui stimulent le système nerveux central, mais une explication complète de leur rôle nécessite une étude détaillée. Des études ex vivo sur des cellules du ganglion cervical supérieur enrichies en NET et OCT3 ont montré qu’en présence de MDPV bloquant les protéines MAT, la méphédrone provoque un efflux de neurotransmetteurs par OCT3, qui est insensible aux effets inhibiteurs du MDPV. La libération de monoamines par OCT3, un transporteur de faible affinité, explique probablement les effets synergiques paradoxaux des inhibiteurs et des substrats.
4. PROFILS NEUROPHARMACOLOGIQUES DES CATHINONES SYNTHÉTIQUES
Les CS peuvent être divisés en trois groupes principaux en fonction de leur puissance inhibitrice relative et de la nature des interactions avec les MAT. Les effets produits par chaque groupe sont comparés aux effets neuropharmacologiques de substances psychoactives classiques et bien étudiées : cocaïne, MDMA, méthamphétamine et pyrovalérone. Ces substances de référence font l’objet de recherches approfondies depuis longtemps et les relations entre leurs propriétés in vitro et leurs effets sur l’organisme ainsi que l’effet clinique induit sont bien connus. En utilisant une approche dans laquelle les CS sont testés en parallèle avec des composés de référence connus, il est possible d’obtenir, sur la base de la similarité obtenue, une bonne estimation de la façon dont les propriétés in vitro des CS se traduisent en propriétés pharmacologiques réelles. La plupart des dérivés de la cathinone présentent une inhibition similaire de la NET, le principal facteur de différenciation étant donc l’interaction avec les systèmes dopaminergiques et sérotoninergiques. La stimulation des systèmes individuels se traduit par des risques spécifiques associés à l’utilisation de ces substances (tableau 2). Les inhibiteurs de SERT sont connus pour être entactogènes et induire un syndrome sérotoninergique. La stimulation noradrénergique entraîne une stimulation cardiaque sympathomimétique, et l’agitation dopaminergique influence fortement le potentiel addictif et les propriétés renforçantes.

Les effets produits par les CS du premier groupe ressemblent à la fois à la MDMA et à la cocaïne. La MDMA stimule la libération de 5-HT tout en inhibant la recapture du SERT et du NET et est moins puissante par rapport au DAT. Elle constitue donc un bon composé de référence pour les CS qui ont un effet particulièrement fort sur les systèmes sérotoninergique et noradrénergique. La cocaïne agit entièrement comme un bloqueur de la recapture, et son action ne s’accompagne donc pas d’un efflux de neurotransmetteurs à travers les MAT. Le blocage se produit avec une puissance similaire pour les trois transporteurs. Par conséquent, le groupe des cathinones dont l’action est similaire à celle de la cocaïne et de la MDMA se distingue par sa capacité à libérer de la 5-HT, ce qui entraîne des signes et des symptômes similaires à ceux observés après l’utilisation de MDMA entactogène, tandis que l’inhibition du recaptage de la DAT et de la NET entraîne également des effets similaires à ceux observés après l’absorption de cocaïne (Simmler et al., 2013, 2014). Les représentants typiques de ce groupe sont la méthylone et la méphédrone, qui, dans des études in vivo de stimulus discriminatoire sur des rats et des singes écureuils, ont complètement remplacé les individus formés à la MDMA. D’autres composés de ce groupe comprennent, entre autres, l’éthylone (1-[1,3-benzodioxol-5-yl]-2-[éthylamino]propan-1-one) et la butylone ([1,3-benzodioxol-5-yl]-2-[méthylamino]butan-1-one).
La méthamphétamine est un substrat pour les MAT et provoque la libération de DA et de NE et, dans une moindre mesure, de 5-HT. Par conséquent, les CS du deuxième groupe, dont l’action est similaire à celle de la méthamphétamine, ont un potentiel addictif élevé en raison d’une forte stimulation dopaminergique et produisent des effets toxiques similaires à ceux de la méthamphétamine et de l’amphétamine. Ces composés sont généralement non substitués sur l’anneau aromatique (ou substitués par du fluor) et ne contiennent pas d’anneau pyrrolidine. Un SC typique ressemblant à la méthamphétamine dans ses effets est son analogue β-cétone : la méthcathinone. La méthcathinone produit une augmentation des niveaux extracellulaires de DA dans le striatum des rongeurs, remplace entièrement les effets du stimulus discriminatif de la méthamphétamine chez les rongeurs et les primates non humains et provoque une augmentation dose-dépendante de l’activité locomotrice horizontale chez les rats. Cet effet est toutefois plus faible que celui des dérivés de la pyrrolidine. Outre la méthcathinone, des dérivés tels que la cathinone, la flephedrone (1-[4-fluorophényl]-2-[méthylamino]propan-1-one), l’éthcathinone (2-éthylamino-1-phényl-propan-1-one), La 3-fluorométhcathinone (1-[3-fluorophényl]-2-[méthylamino]propan-1-one) et la buphedrone (2-[méthylamino]-1-phénylbutan-1-one) appartiennent à ce groupe.
Le troisième groupe de CS, qui se distingue par la présence d’un cycle pyrrolidine, a une action comparable à celle de la pyrovalérone (1-[4-méthylphényl]-2-[1-pyrrolidinyl]pentan-1-one), un dérivé de la cathinone déjà connu depuis les années 1960. Les CS de ce groupe sont des bloqueurs extrêmement puissants du DAT et du NET, tout en ayant une faible affinité pour le SERT. En outre, ils sont considérés comme totalement incapables de libérer des monoamines. Cependant, de récentes études in vitro examinant l’efflux de monoamines radiomarquées dans des lignées cellulaires monoclonales de rein embryonnaire humain (HEK) enrichies en NET humaines suggèrent que certains des CS, en particulier les dérivés a-PPP, peuvent induire la libération de NE par le biais de la NET en agissant comme des agents de libération partielle. Les pyrovalérones sont très lipophiles et des études dans des modèles de barrière hémato-encéphalique (BHE) in vitro ont montré qu’elles traversent la BHE plus efficacement que des dérivés moins lipophiles. Cependant, des études in vivo plus récentes sur des tissus cérébraux de rats démontrent la relation inverse et, en général, ce sont les composés plus polaires qui traversent la BHE plus efficacement. Néanmoins, les CS contenant un anneau de pyrrolidine, probablement en raison de leur lipophilie, et en raison d’une puissance élevée contre le DAT et le NET, montrent une activité élevée déjà à très faible dose, ce qui est en corrélation avec les doses plus faibles appliquées rapportées par les utilisateurs par rapport à d’autres CS. Il a été démontré que les CS de ce groupe stimulent fortement l’activité locomotrice des rongeurs, médiée par le récepteur D1 de la dopamine, et provoquent une augmentation des niveaux de DA mais, remarquablement, aussi de 5-HT dans le striatum de la souris. Des études d’affinité in vitro indiquent que les pyrovalérones ne devraient pas interagir avec le SERT et ne devraient pas provoquer la libération de 5-HT, de sorte que la concentration élevée de 5-HT observée peut vraisemblablement s’expliquer par le couplage fonctionnel des systèmes dopaminergiques et sérotoninergiques. En cas de fortes concentrations extracellulaires de DA, on observe le phénomène de sa captation par le SERT, accompagné d’une libération concomitante de 5-HT médiée par le SERT. Il est intéressant de noter que des études récentes chez la souris ont montré que les CS possédant un cycle pyrrolidine, contrairement aux CS du deuxième groupe de type méthamphétamine, provoquent, en plus de l’activité locomotrice horizontale, également une activité locomotrice verticale accrue en fonction de la dose. L’augmentation de l’activité locomotrice verticale est probablement le signe d’une stimulation dopaminergique particulièrement puissante et sélective et d’une réduction de l’anxiété, contrairement aux dérivés tels que ceux du deuxième groupe de type méthamphétamine. Les composés populaires de ce groupe comprennent, entre autres, le PV8 (1-Phényl-2-pyrrolidine-1-ylheptan-1-one), le MDPV et l’α-PVP (1-phényl-2-[1-pyrrolidinyl]-1-pentanone).
5. DÉTERMINANTS STRUCTURELS DE L’ACTIVITÉ DES CATHINONES SYNTHÉTIQUES
Les effets des CS sont évalués et prédits à l’aide de la méthode des relations structure-activité (SAR). Ces études se concentrent sur des changements structurels uniques pour observer les tendances et les relations dans les changements d’activité vis-à-vis des protéines sélectionnées. La complexité des études SAR est influencée par les différences dans la nature et la force des interactions indépendantes des CS avec trois protéines MAT. Certains CS agissent sélectivement sur un ou deux transporteurs seulement tout en étant inactifs sur les autres, tandis que d’autres présentent un caractère hybride en agissant comme inhibiteur du DAT et substrat du SERT.
Les facteurs les plus importants qui déterminent si un dérivé agit comme un substrat ou un bloqueur sont la taille et le nombre des substituants aminés. Plus la taille et le nombre des substituants aminés augmentent, plus la puissance de l’action bloquante augmente. Les amines tertiaires et les amines secondaires avec des substituants volumineux présentent une forte inhibition des MAT et ne stimulent pas la libération de neurotransmetteurs dans la fente synaptique. En revanche, les amines primaires ou les amines secondaires avec de petits substituants agissent principalement comme des substrats et provoquent simultanément la libération de monoamines. Cependant, l’augmentation de la taille des substituants aminés ne conduit pas toujours à une augmentation de la puissance inhibitrice. Lors d’études sur le recaptage des synaptosomes de rats à l’aide de radioligands, il a été démontré que l’inhibition in vitro du recaptage de la DA par le DAT humain est plus forte lorsque le groupe N-méthyle est remplacé par un groupe N-éthyle, mais qu’elle diminue lorsque la partie pyrrolidine est remplacée par un anneau pipéridine ou un groupe diéthyle plus encombrant. En outre, ces observations se sont révélées cohérentes avec les études d’amarrage moléculaire. L’allongement de la chaîne alkyle latérale au niveau du carbone α est un autre facteur qui influe sur le pouvoir inhibiteur. Des essais de liaison et d’absorption de radioligands utilisant des lignées cellulaires HEK transfectées avec des MAT humaines ont montré que l’allongement de la chaîne latérale d’un à cinq atomes de carbone augmente l’inhibition de la DAT jusqu’à 100 fois. Il convient toutefois de noter que des études in vivo sur l’activité locomotrice ont montré que l’allongement de la chaîne latérale n’augmente la puissance du SC chez la souris que jusqu’à un certain point. Les PV8 et PV9, qui ont respectivement des chaînes latérales de 5 et 6 carbones, présentent une puissance plus faible que l’α-PVP à 3 carbones, comme le confirment les rapports des utilisateurs de ces substances, et un tel effet peut probablement être dû à des propriétés pharmacocinétiques différentes des dérivés plus lipophiles et plus volumineux (Wojcieszak et al., 2018). Il est intéressant de noter que ces dernières années, la plupart des CS les plus populaires ont une chaîne latérale de carbone allongée.
L’un des CS de “première génération” les plus populaires était le MDPV, un bloqueur puissant qui se distinguait par la présence d’un anneau de pyrrolidine. En 2013, le MDPV a été “déconstruit” afin d’évaluer quels substituts et dans quelle mesure déterminent le pouvoir inhibiteur sur la DAT. À cette fin, l’expérience électrophysiologique de voltage clamp en réponse au MDPV et à ses dérivés a été utilisée avec des ovocytes de Xenopus laevis exprimant le DAT humain, ainsi qu’un essai d’inhibition de l’absorption des monoamines radiomarquées avec des cellules HEK enrichies en DAT humain. Les études ont montré que la privation d’un dérivé du groupe β-cétone entraîne une diminution de plusieurs fois de la puissance inhibitrice vis-à-vis de la DAT et une augmentation de la puissance vis-à-vis de la SERT, ce qui est cohérent avec l’observation selon laquelle les analogues amphétaminiques des CS sont des inhibiteurs moins puissants de la DAT. Dans le cas des pyrovalérones, la présence du groupe 3,4-méthylènedioxy a un effet négligeable sur leur puissance vis-à-vis de la DAT. Sans ce groupe, l’α-PVP est un bloqueur de DAT in vitro aussi puissant que le MDPV. D’autre part, la longueur de la chaîne alkyle au niveau du carbone α a un impact considérable sur le pouvoir inhibiteur du dérivé. Le raccourcissement de la chaîne latérale du MDPV de 2 atomes de carbone entraîne une réduction de la puissance de plus de 25 fois. Le remplacement de l’anneau pyrrolidine par un groupe N,N-diméthyle moins volumineux réduit le pouvoir inhibiteur d’environ 5 fois. La réduction du nombre de substituants aminés de tertiaire à secondaire et primaire réduit également de manière significative le pouvoir inhibiteur de la DAT. Il convient toutefois de noter que les alkylamines primaires à longue chaîne ainsi que les alkylamines tertiaires à courte chaîne continuent d’agir comme des bloqueurs de la MAT, bien que l’effet le plus fort soit démontré par les amines tertiaires ayant simultanément une chaîne latérale de carbone étendue. Inversant cette relation, les dérivés sans N-substituants et sans chaîne latérale de carbone étendue agissent comme des substrats de la MAT.
Malgré sa grande utilité, il convient d’être prudent lorsque l’on tire des conclusions des études SAR. Ces études sont réalisées in vitro ou sur des rongeurs, ce qui peut interférer avec la traduction directe des résultats de la recherche dans l’action réelle de ces drogues dans le corps humain. Il est également important de garder à l’esprit l’impact des interactions avec d’autres protéines telles que les VMAT et les OCT. En outre, les études peuvent ne pas prendre en compte les facteurs pharmacocinétiques affectant l’activité de ces composés, tels que l’absorption, le métabolisme et la capacité à traverser la BHE.
Un paramètre utile pour comparer le mode d’action des CS est le ratio de sélectivité relative de la puissance d’inhibition de la captation du DAT par rapport à la puissance d’inhibition de la captation du SERT (tableau 3). Le rapport DAT/SERT, calculé comme suit : 1/CIC50 DAT:1/CIC50 SERT, prend une large gamme de valeurs, les valeurs élevées indiquant une plus grande sélectivité pour la DAT et les valeurs faibles pour la SERT. Un coefficient de sélectivité DAT/SERT <0,1 indique un effet entactogène de type MDMA (DAT/SERT de la MDMA = 0,08), et une valeur élevée de ce coefficient (>10) est associée à un fort potentiel de dépendance et à un effet psychostimulant de type méthamphétamine (DAT/SERT de la méthamphétamine = 22). La valeur la plus élevée de ce coefficient se retrouve chez les pyrovalérones fortement addictives, où il peut atteindre plusieurs milliers. Le coefficient DAT/SERT est proche de un dans les CS présentant un potentiel de dépendance modéré et des effets similaires à ceux de la cocaïne (DAT/SERT de la cocaïne = 3,1).
[TABLEAU 3]
Dans certains cas, le rapport DAT/SERT a une applicabilité limitée car certains dérivés peuvent présenter des propriétés différentes pour des valeurs comparables du rapport DAT/SERT. Les CS présentant des valeurs similaires peuvent avoir des caractéristiques de performance substrat/bloquant différentes. Les analogues de la méthylone qui diffèrent par la longueur de leur chaîne alkyle présentent des rapports DAT/SERT similaires, mais l’allongement de la chaîne fait que les substrats de la DAT changent de nature et deviennent des bloquants tout en conservant leur activité en tant que substrats de la SERT. La pentylone et la butylone induisent toutes deux une augmentation des concentrations in vivo de DA et de 5-HT dans l’espace extracellulaire du noyau accumbens chez le rat. Cependant, la pentylone agit principalement comme un bloqueur de la DAT et entraîne une augmentation des concentrations de DA par rapport à celles de 5-HT, tandis que la butylone, dont la chaîne latérale est plus courte d’un atome de carbone, est principalement un substrat puissant de la SERT et entraîne principalement une augmentation de la 5-HT par rapport à celle de la DA. Par conséquent, les effets de la pentylone sont similaires à ceux de la méthamphétamine dopaminergique et, chez les rats, présentent une plus forte stimulation de l’activité locomotrice, tandis que les effets de la butylone sont plus similaires à ceux de la MDMA entactogène et présentent moins d’effets renforçants.
La stéréoisomérie a également un impact majeur sur l’action des CS. Dans le cas des CS dotés d’un cycle pyrrolidine, comme le MDPV et l’α-PVP, leurs stéréoisomères S sont plusieurs fois plus puissants que les stéréoisomères R. Dans des études sur le rat, le (S)-α-PVP a montré, à des doses 30 fois inférieures au (R)-α-PVP, la même augmentation de l’activité locomotrice et des effets cardiovasculaires et a démontré, dans des études de microdialyse, la même augmentation de la concentration extracellulaire de dopamine dans le noyau accumbens. En conséquence, des différences tout aussi importantes ont été démontrées pour les énantiomères du MDPV, où le (S)-MDPV a provoqué une activité locomotrice beaucoup plus forte chez les rats, ainsi qu’une augmentation de la pression artérielle et de la fréquence cardiaque que le (R)-MDPV. En outre, une différence similaire dans l’essai d’auto-administration selon un calendrier de ratio progressif s’est produite dans la puissance entre le (S)-MDPV et le (R)-MDPV. L’impact de la stéréochimie est plus complexe pour les substances qui sont des substrats de transporteurs. Il affecte non seulement leur puissance, mais aussi la sélectivité de l’inhibition de la DAT/SERT. Dans le cas de la méphédrone, les stéréoisomères ne présentent pas de grande différence de puissance en tant que substrats de la DAT ; cependant, l’isomère R est sélectif pour la DAT, tandis que l’isomère S ne présente pas une telle sélectivité et interagit également en tant que substrat du SERT. La sélectivité de la (R)-méphedrone et son absence de propriétés sérotoninergiques se traduisent par un profil plus stimulant que celui de la (S)-méphedrone, ce qui, pour le stéréoisomère R, s’est traduit dans les études sur le rat par une plus grande activité locomotrice, une préférence de place conditionnée affichée et une plus grande facilitation de l’autostimulation intracrânienne que pour le stéréoisomère S. Néanmoins, il convient de noter que les études SAR sont principalement réalisées sur des mélanges racémiques de CS, car c’est sous cette forme qu’ils sont vendus et utilisés.
Pour les libérateurs de type substrat, le type de substituant au niveau du cycle aromatique, sa position et sa capacité à retirer des électrons sont très importants. Le rapport de sélectivité DAT/SERT peut être contrôlé par la position des substituants. Des études in vitro de dosage de radioligands sur des synaptosomes de rat ont montré que les dérivés para-substitués sont beaucoup plus puissants sur SERT que leurs homologues ortho- et méta-substitués. Les dérivés ortho-substitués présentent principalement une activité dopaminergique, les dérivés méta-substitués présentent des rapports DAT/SERT plus faibles, et les dérivés para-substitués se caractérisent par une activité sérotoninergique. Le rapport de sélectivité DAT/SERT pour l’éphédrone non substituée est supérieur à 300, tandis que la méthédrone ayant un substituant méthoxy en position para interagit quatre fois plus fortement avec le SERT qu’avec le DAT, présentant ainsi vraisemblablement des effets entactogènes similaires à ceux de la MDMA. Les dérivés disubstitués et les dérivés avec des substituants stériquement étendus au niveau du cycle phényle favorisent l’interaction avec SERT. Pour les CS sans groupe pyrrolidine, la substitution du cycle aromatique par un groupe 3,4-méthylènedioxy favorise l’inhibition du SERT. Cependant, l’effet résultant n’est pas toujours significatif, car la pentylone et son analogue dépourvu du groupement 3,4-méthylènedioxy, la pentedrone, ne présentent chez le rat qu’une faible différence au niveau des effets comportementaux, comme l’activité locomotrice, et des effets de renforcement déterminés à l’aide de la technique d’auto-administration par voie intraveineuse. D’autre part, les dérivés de la pyrovalérone avec le groupe 3,4-méthylènedioxy- ne montrent qu’une capacité in vitro légèrement supérieure à inhiber le SERT que ceux sans ce groupe.
En conclusion, les études SAR constituent un outil précieux pour évaluer les profils toxicologiques des CS. Le grand nombre de composés possibles fait que les études systémiques sur la relation structure-activité sont d’une grande aide pour prédire et évaluer les propriétés des composés nouvellement développés. Le type et la force des interactions entre les CS et une protéine particulière se traduisent par les effets, les signes et symptômes et le type d’empoisonnement. Les relations trouvées permettent de comparer les mécanismes d’action et les toxicités de dérivés similaires de la cathinone et de composés de référence en se basant uniquement sur la structure.
