
Heal DJ, Smith SL, Gosden J, Nutt DJ. Amphetamine, past and present–a pharmacological and clinical perspective. J Psychopharmacol. 2013 Jun;27(6):479-96. doi: 10.1177/0269881113482532. Epub 2013 Mar 28. PMID: 23539642; PMCID: PMC3666194.
Abstract.
L’amphétamine a été découverte il y a plus de 100 ans. Depuis lors, elle est passée d’un médicament en vente libre sans ordonnance comme panacée pour un large éventail de troubles à un médicament contrôlé très restreint dont les applications thérapeutiques se limitent au trouble déficitaire de l’attention avec hyperactivité (TDAH) et à la narcolepsie. Cette étude décrit la relation entre la structure chimique et la pharmacologie de l’amphétamine et de ses congénères. Les diverses actions pharmacologiques de l’amphétamine se traduisent non seulement par une efficacité thérapeutique, mais aussi par la production d’effets indésirables et la responsabilité de l’abus récréatif. En conséquence, l’équilibre entre les bénéfices et les risques est le principal défi à relever pour son utilisation clinique. L’étude présente les progrès du développement pharmaceutique depuis l’introduction des formulations d’amphétamine à prise unique quotidienne jusqu’à la lisdexamfétamine, premier promédicament de d-amphétamine approuvé pour la prise en charge du TDAH chez les enfants, les adolescents et les adultes. La voie métabolique inhabituelle par laquelle la lisdexamfétamine délivre la d-amphétamine apporte une contribution importante à sa pharmacologie. La façon dont le profil pharmacocinétique/pharmacodynamique particulier de la lisdexamfétamine se traduit par une efficacité soutenue dans le traitement du TDAH et par un potentiel réduit d’abus récréatif est également abordée.
Une courte histoire de l’amphétamine.
Bien que l’α-méthylphénéthylamine (amphétamine) racémique ait été découverte par Barger et Dale en 1910, ce n’est qu’en 1927 que cette molécule a été synthétisée pour la première fois par le chimiste G. A. Alles, alors qu’il cherchait un substitut moins coûteux et plus facile à synthétiser pour l’éphédrine. Les expériences menées sur des animaux et des sujets humains par Alles et d’autres ont révélé sans équivoque la capacité de l’α-méthylphénéthylamine à inverser l’anesthésie induite par la drogue et à produire de l’excitation et de l’insomnie. Le nom commercial “Benzedrine®” pour l’α-méthylphénéthylamine racémique a été enregistré par la société pharmaceutique Smith, Kline and French. L'”amphétamine”, qui est le nom générique de la benzédrine conçu par le Conseil de la pharmacie et de la chimie de l’Association médicale américaine, n’a été adopté que de nombreuses années plus tard. C’est la raison pour laquelle le nom Benzedrine, et non amphétamine, apparaît dans toutes les premières publications. Smith, Kline et French ont mis la benzédrine sur le marché en 1935 comme traitement de la narcolepsie (pour laquelle elle est encore utilisée aujourd’hui), de la dépression légère, du parkinsonisme post-encéphalitique et d’un grand nombre d’autres troubles.
En tant que molécule à centre chiral unique, l’amphétamine existe sous deux formes optiquement actives, à savoir les isomères dextro (ou d-) et lévogyre (ou l-) ou énantiomères (figure 1). Smith, Kline et French ont synthétisé les deux isomères et, en 1937, ont commencé à commercialiser la d-amphétamine, le plus puissant des deux isomères, sous le nom de Dexedrine®. La vente de Benzedrine et de Dexedrine dans les pharmacies était libre jusqu’en 1939, date à laquelle ces médicaments ne pouvaient être obtenus que sur ordonnance d’un médecin agréé ou en signant le registre des poisons. Les propriétés d’amélioration cognitive de l’amphétamine ont été rapidement reconnues, les rapports faisant état d’une amélioration des tests d’intelligence par la benzédrine, ce qui a conduit à son utilisation généralisée pour réduire le stress et améliorer la concentration et les performances intellectuelles des universitaires, des étudiants et des professionnels de la santé. Dans sa revue de 1946, Bett commente l’utilisation répandue de “pilules énergétiques” par les forces alliées pendant la Seconde Guerre mondiale, estimant que 150 millions de comprimés de benzédrine ont été fournis au personnel militaire britannique et américain au cours du conflit mondial. Malgré une couverture considérable dans la littérature médicale et la presse populaire décrivant les puissants effets centraux de ces nouvelles drogues, le potentiel de dépendance de l’amphétamine a été largement ignoré.

C’est Bradley (1937) qui a rapporté pour la première fois les effets bénéfiques de la benzédrine dans le traitement d’enfants présentant de graves problèmes de comportement, que l’on diagnostiquerait aujourd’hui comme souffrant d’un trouble déficitaire de l’attention/hyperactivité (TDAH). Bradley a traité 30 sujets pendant une semaine et a observé chez environ la moitié d’entre eux des améliorations remarquables de leurs résultats scolaires, de leur comportement et de leur attitude. Ces bénéfices thérapeutiques provenaient sans équivoque du médicament puisqu’ils étaient apparents dès le premier jour du traitement à la benzédrine et disparaissaient dès l’arrêt de celui-ci. Bien que la l-amphétamine (Cydril®) ait suscité beaucoup moins d’intérêt que le racémate ou l’isomère d, les essais cliniques menés dans les années 1970 ont démontré que les deux isomères de l’amphétamine étaient cliniquement efficaces pour traiter le TDAH. L’utilisation de la benzédrine pour traiter le TDAH a considérablement diminué après que Gross (1976) a rapporté que le racémate était beaucoup moins efficace cliniquement que la dexédrine. Actuellement, la seule utilisation de la l-amphétamine dans les médicaments contre le TDAH est l’amphétamine à sels mixtes/énantiomères mixtes (MES-amphétamine), qui consiste en un mélange énantiomérique 3:1 de sels de d-amphétamine et de l-amphétamine, disponible sous forme de formulations à libération immédiate (Adderall®, générique) et à libération prolongée (Adderall XR®, générique). Un développement récent dans le domaine des amphétamines est l’introduction d’un promédicament de l’amphétamine, le dimésylate de lisdexamfétamine (Vyvanse®). La lisdexamfétamine est un acide aminé naturel, la L-lysine, lié de manière covalente à la d-amphétamine par l’intermédiaire d’un groupe amide. Il a été approuvé pour la prise en charge du TDAH chez les enfants (âgés de 6 à 12 ans), les adolescents et les adultes aux États-Unis et au Canada. Il est actuellement en cours de développement pour une utilisation clinique dans le traitement du TDAH dans un certain nombre de pays européens. La voie métabolique de la lisdexamfétamine est inhabituelle car, après absorption dans la circulation sanguine, elle est métabolisée par les globules rouges en d-amphétamine et en L-lysine, un acide aminé naturel, par hydrolyse enzymatique à vitesse limitée. Le tableau 1 donne un aperçu des médicaments à base d’amphétamine.
[TABLEAU 1]
Une perspective clinique sur l’utilisation de l’amphétamine dans le traitement du TDAH.
Le TDAH est sans doute le trouble psychiatrique le plus sous-diagnostiqué et le plus mal traité, en particulier chez les adultes. Les données européennes les plus récentes suggèrent qu’environ 5 % de la population souffre de TDAH au cours d’une année, soit un total d’environ 3 millions de patients en Europe. Selon d’autres estimations, le coût de chaque patient s’élève à environ 5 000 £ par an au Royaume-Uni. Sur ce total, un peu plus de la moitié correspond à des coûts de traitement directs et le reste à des coûts indirects, par exemple la perte de productivité, le préjudice social, l’impact négatif sur la vie familiale, l’augmentation de l’incidence des accidents et les coûts associés à la criminalité et à l’intervention de la justice. L’impact en termes de perte de qualité de vie (jours vécus avec un handicap) place le TDAH dans le top 10 des troubles du cerveau en Europe. Le traitement du TDAH est généralement inadéquat, les estimations suggérant que, dans le meilleur des cas, moins d’un tiers des patients diagnostiqués reçoivent un traitement approprié.
Bien que l’amphétamine ait été établie comme un traitement efficace du TDAH, ainsi que d’autres troubles du système nerveux central (CNS) tels que la narcolepsie depuis des décennies, son utilisation au Royaume-Uni (et dans le contexte européen plus large) a été plutôt limitée par rapport à son utilisation généralisée aux États-Unis. Les raisons en sont complexes et tiennent aux attitudes sociales et médicales à l’égard du TDAH, aux politiques de commercialisation de l’industrie pharmaceutique, ainsi qu’aux préoccupations concernant l’utilisation de médicaments dans des indications pédiatriques qui sont perçus comme ayant un fort potentiel d’abus récréatif et de toxicomanie.
Le TDAH a longtemps souffert d’être considéré comme un diagnostic “américain” et, pendant de nombreuses décennies, certains experts en pédopsychiatrie ont tenté de nier, ou du moins de minimiser, son existence au Royaume-Uni. En outre, dans les rares cas où le trouble était identifié, l’option thérapeutique privilégiée était la psychothérapie, car elle correspondait à la formation des pédopsychiatres et des psychologues responsables de la prise en charge de ces patients. Il revenait à certains pédiatres de développer l’expertise nécessaire dans l’utilisation des stimulants pour traiter les enfants atteints de TDAH, ce que nombre d’entre eux ont fait avec succès. Ces dernières années, les pédopsychiatres ont commencé à jouer un rôle de prescripteurs, en utilisant principalement des préparations à base de méthylphénidate.
Les amphétamines, c’est-à-dire l’amphétamine racémique, la d-amphétamine et la méthamphétamine, ont été largement utilisées pour favoriser l’éveil pendant la Seconde Guerre mondiale, ce qui a entraîné une forte augmentation de la production qui s’est traduite par d’importants excédents de ces drogues après la guerre. Une grande partie de ces stocks s’est retrouvée sur le “marché noir” et, dans les années 1950, l’abus de d-amphétamine a été reconnu. Dans une étude classique de cette période, Connell, de l’Institut de psychiatrie, a fait état d’un groupe de gros consommateurs de d-amphétamine qui étaient devenus paranoïaques. Cela a mis en évidence les dangers psychiatriques potentiels de ce médicament et a peut-être encouragé les prescripteurs à délaisser la d-amphétamine au profit du méthylphénidate. Un autre facteur a été l’utilisation de la d-amphétamine comme antidépresseur dans les années 50, avant la découverte des inhibiteurs tricycliques de la recapture des monoamines. Il y a eu des cas d’utilisation abusive par des patients, ainsi qu’un degré important de détournement du médicament prescrit pour une utilisation abusive par des jeunes, ce qui peut avoir contribué à la méfiance des prescripteurs à l’égard de son utilisation clinique. Plus tard, des flambées locales d’abus de d-amphétamine se sont produites dans diverses régions du Royaume-Uni, souvent à partir de d-amphétamine synthétisée localement ; là encore, les médecins ont hésité à prescrire de la d-amphétamine de peur de contribuer à son usage abusif. Aux États-Unis, les médicaments contenant de la d-amphétamine, en particulier la MES-amphétamine, ont été très largement utilisés dans le traitement du TDAH. La familiarité avec les amphétamines prescrites et la disponibilité croissante de formulations de plus en plus inviolables pour réduire le risque d’abus, comme l’Adderall XR®, ont créé une situation où, aux États-Unis, le risque d’abus de d-amphétamine est perçu comme étant similaire à celui du méthylphénidate. Ce fait, ainsi que la perception que la d-amphétamine est beaucoup plus sûre que la méthamphétamine, un stimulant plus puissant et plus durable, dont l’abus est aujourd’hui très répandu, ont conduit les médecins américains à adopter une attitude plus détendue en ce qui concerne la prescription de d-amphétamine. Heureusement, pour des raisons obscures, l’abus récréatif de méthamphétamine n’a jamais vraiment pris en Europe, et la quasi-totalité de l’usage illégal des amphétamines se limite à la d-amphétamine sous forme de sel de sulfate.
La pharmacologie de l’amphétamine.
La structure chimique, en particulier la structure tridimensionnelle (3-D) de l’amphétamine, est essentielle pour déterminer les effets pharmacologiques qui sous-tendent ses avantages thérapeutiques considérables et son risque d’abus récréatif. L’amphétamine appartient à la classe des médicaments appelés “β-phényléthylamines”. Bien qu’elle ait été synthétisée plusieurs décennies avant la découverte que les monoamines, c’est-à-dire la noradrénaline (norépinéphrine), la dopamine et la 5-hydroxytryptamine (5-HT ; sérotonine), étaient des neurotransmetteurs majeurs dans les systèmes nerveux central et périphérique, la raison de la synthèse de l’amphétamine racémique était en partie sa similarité structurelle avec la molécule biologiquement active, l’éphédrine.
Comme le montre la figure 1, la similitude entre les structures chimiques des neurotransmetteurs catécholamines, noradrénaline et dopamine, et les isomères de l’amphétamine est tout à fait évidente. Les structures tridimensionnelles des molécules de catécholamines et d’amphétamine révèlent la longue conformation planaire commune à tous ces composés. Pour les isomères de l’amphétamine, c’est leur conformation plane, leur taille moléculaire similaire à celle des monoamines, la présence d’un cycle aromatique et d’un azote sur la chaîne latérale arylique qui sont les propriétés physico-chimiques préalables d’un substrat compétitif pour les transporteurs de recapture des monoamines, à savoir le NET (transporteur de noradrénaline), le DAT (transporteur de dopamine) et le SERT (transporteur de 5-HT).
La figure 2 illustre le mécanisme responsable du transport de la recapture des monoamines et de l’amphétamine dans les terminaux nerveux présynaptiques. Une molécule de neurotransmetteur monoaminergique ou d’amphétamine s’associe à deux ions Na+ et un ion Cl-, et le complexe moléculaire qui en résulte est activement transporté dans le terminal présynaptique par le transporteur de recapture des monoamines correspondant. La force motrice de ce mécanisme de transport actif est un gradient de concentration d’ions Na+ (Na+ élevé à l’extérieur du terminal nerveux / Na+ faible à l’intérieur). Le gradient de concentration de Na+ est maintenu par la Na+/K+ ATPase qui pompe deux ions Na+ hors de la cellule tout en pompant simultanément un ion K+. Il existe deux réservoirs de neurotransmetteurs monoaminergiques dans chaque type de terminal nerveux : le réservoir cytosolique qui contient les monoamines nouvellement synthétisées et le réservoir vésiculaire qui stocke les monoamines et à partir duquel elles sont libérées lorsque les neurones déclenchent des potentiels d’action.
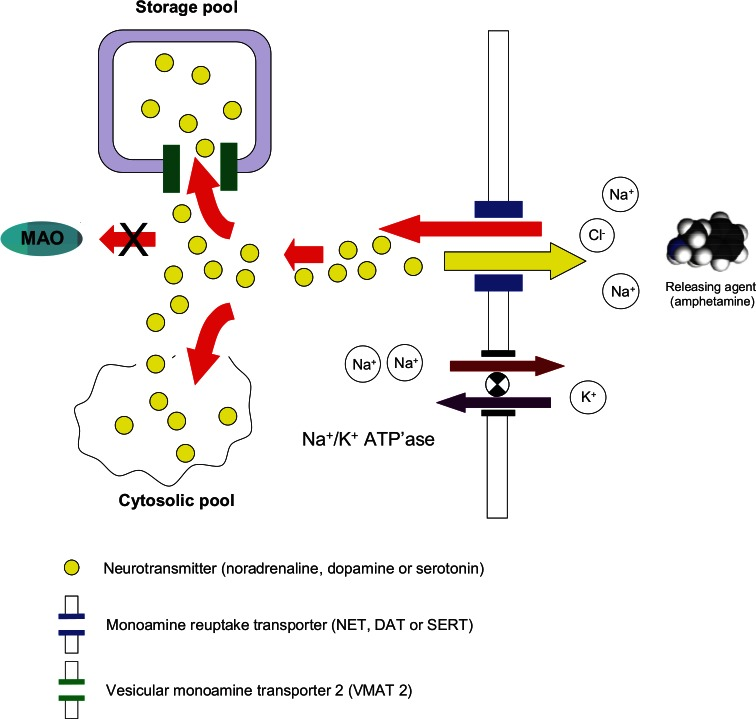
Bien que la concentration d’un neurotransmetteur monoaminergique dans le cytosol du terminal nerveux présynaptique soit régulée par ses taux de synthèse, de libération, de recapture et de catabolisme, il est désormais reconnu que le transport de la monoamine dans les granules de stockage vésiculaires joue un rôle crucial dans ce processus. La translocation des monoamines du pool cytosolique vers le pool de stockage est assurée par un système de transport actif similaire, le transporteur vésiculaire de monoamines 2 (VMAT2). Étant donné que l’amphétamine entre en compétition avec les monoamines endogènes pour le transport dans les terminaux nerveux via le NET, le DAT ou le SERT, plus la concentration d’amphétamine présente dans la synapse est élevée, plus le nombre de molécules d’amphétamine transportées par rapport à chaque molécule de monoamine est important (voir la figure 3). Une fois dans le terminal présynaptique, l’amphétamine déplace les monoamines du pool cytosolique. En outre, comme l’amphétamine a également une affinité pour la VMAT2, elle empêche la translocation des monoamines dans les vésicules de stockage intraneuronales. Ces actions ont pour effet d’inverser la direction du transporteur de recapture, de sorte qu’au lieu de pomper le neurotransmetteur de la synapse vers le terminal nerveux, il pompe le neurotransmetteur des neurones vers la synapse. Ce processus est appelé “transport inverse” ou “rétro-transport”.
Conformément au mécanisme décrit ci-dessus, des expériences in vitro ont démontré sans équivoque que les isomères d et l de l’amphétamine libèrent de manière non sélective des [3H]monoamines à partir de tranches préchargées ou de synaptosomes préparés à partir de cerveau de rat. Des rapports expérimentaux indiquent que la d-amphétamine libère de la [3H] noradrénaline, de la dopamine et de la 5-HT à partir de synaptosomes et de tranches de cerveau. La l-amphétamine libère de la noradrénaline, de la dopamine et de la 5-HT à partir des synaptosomes et de la noradrénaline et de la dopamine à partir de tranches de cerveau de rat. En comparant les puissances relatives de la d- et de la l-amphétamine, Heikkila et al. (1975) et Easton et al. (2007) ont rapporté que l’isomère d était environ quatre fois plus puissant que l’isomère l en tant que libérateur de [3H]dopamine. En revanche, la l-amphétamine était aussi puissante, voire plus, que la d-amphétamine pour libérer la [3H]noradrénaline. Les transporteurs de monoamines ne sont pas particulièrement sélectifs quant aux monoamines qu’ils transportent, et ce manque de sélectivité s’explique par leur étroite similarité structurelle (figure 1). De plus, cette similarité structurelle entre les neurotransmetteurs monoaminergiques et l’amphétamine explique pourquoi cette dernière a une action promiscuité pour libérer les monoamines importantes du CNS (noradrénaline, dopamine et 5-HT). L’amphétamine libère également de l’adrénaline à partir du système nerveux sympathique périphérique, une action liée à ses effets secondaires cardiovasculaires. Bien que la plupart de ces expériences aient porté sur les effets des isomères de l’amphétamine sur la libération basale de [3H]monoamine à partir de synaptosomes ou de tranches, l’amphétamine augmente également l’efflux stimulé électriquement (Easton et al., 2007). Cette action indique que son mécanisme de rétro-transport peut agir à la fois en coopération et indépendamment de l’excitation neuronale.
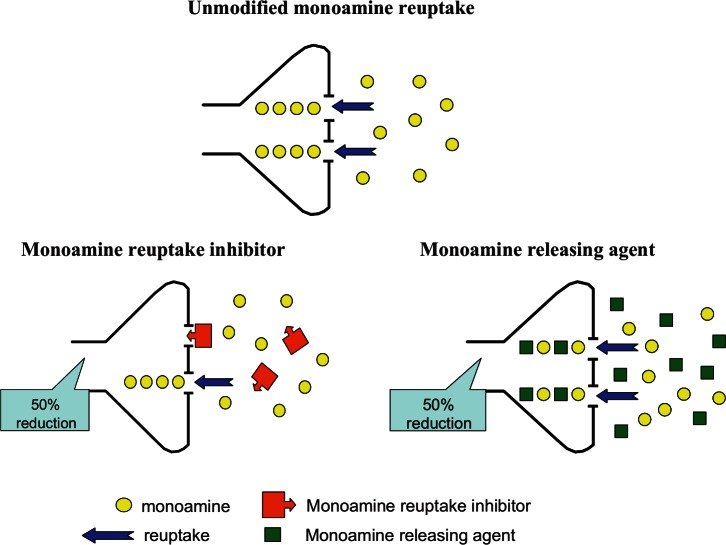
Bien que l’effet pharmacologique de l’amphétamine soit principalement médié par la libération de monoamines, ce mécanisme est complété par l’inhibition de la recapture et probablement aussi par l’inhibition de la monoamine oxydase (MAO), qui se combinent de manière additive ou synergique pour augmenter les concentrations synaptiques de monoamines. La description de l’amphétamine comme “inhibiteur de la recapture des monoamines” est souvent source de confusion, et la différence entre les mécanismes de l’amphétamine, qui est un substrat compétitif du transport de la recapture, et les inhibiteurs classiques de la recapture est illustrée dans la figure 3. La puissance des isomères de l’amphétamine en tant qu’inhibiteurs de la recapture des monoamines est résumée dans le tableau 2 et comparée à celle de certains inhibiteurs classiques de la recapture très puissants. Il est généralement admis que la d-amphétamine est un faible inhibiteur de la recapture de la dopamine avec une valeur Ki d’environ 100 nM, un inhibiteur modérément puissant de la recapture de la noradrénaline (Ki = 40-50 nM) et un très faible inhibiteur de la recapture de la 5-HT (Ki = 1,4-3,8 µM). Les comparaisons des isomères de l’amphétamine révèlent que la l-amphétamine est 3,2 à 7 fois moins puissante que la d-amphétamine en tant qu’inhibiteur de la recapture de la dopamine, mais qu’elle n’est que 1,8 fois moins puissante contre la noradrénaline. Sa puissance est si faible que la l-amphétamine ne serait pas considérée comme un inhibiteur de la recapture de la 5-HT.
Enfin, les monoamines en excès dans le terminal nerveux sont catabolisées par la MAO, une enzyme liée à la mitochondrie. L’inhibition de la MAO augmenterait encore la quantité de neurotransmetteur disponible pour le rétro-transport dans la synapse. Les isomères de l’amphétamine sont connus depuis longtemps pour être des inhibiteurs de cette importante enzyme de catabolisme. Bien que ce mécanisme soit souvent écarté parce que l’amphétamine est un inhibiteur relativement faible de la MAO, il est probable qu’une certaine inhibition de cette enzyme se produise dans la situation où l’amphétamine est concentrée dans les terminaisons nerveuses présynaptiques, comme le montre la figure 3.
Bien que les expériences in vitro donnent un bon aperçu des mécanismes individuels, l’efficacité de l’amphétamine par rapport à d’autres agonistes monoaminergiques indirects, par exemple les inhibiteurs classiques du recaptage, ne peut être estimée qu’à partir d’expériences in vivo. Nous avons utilisé la microdialyse intracérébrale à double sonde pour explorer les effets in vivo de la d- et de la l-amphétamine chez le rat spontanément hypertendu (SHR), qui a été proposé comme modèle rongeur du TDAH.
Les deux isomères de l’amphétamine ont augmenté de manière dose-dépendante les concentrations extracellulaires de noradrénaline dans le cortex préfrontal (PFC) et de dopamine dans le striatum. La pharmacodynamique de leurs effets est typique de ceux rapportés pour les agents libérant des monoamines, c’est-à-dire un début d’action rapide avec des augmentations maximales de l’efflux de noradrénaline et de dopamine survenant entre 30 et 45 minutes, des effets importants (400-450% de la ligne de base pour la noradrénaline et 700-1500% de la ligne de base pour la dopamine), avec un déclin relativement rapide après le maximum (Figure 4). Bien qu’aucun résultat comparatif n’ait été inclus dans cette étude, l’ampleur des augmentations produites par les isomères de l’amphétamine est plus importante que celles rapportées pour les inhibiteurs classiques de la recapture tels que l’atomoxétine ou le bupropion, et il n’y a pas de plafond dose-effet pour les actions de l’amphétamine. Lorsque l’on compare les effets des médicaments sur l’efflux de catécholamines dans le PFC, il est important de tenir compte de la neuroanatomie très inhabituelle de cette région cérébrale. La densité des sites DAT sur les neurones dopaminergiques du PFC est très faible et, par conséquent, la majeure partie de la dopamine libérée est séquestrée via le NET dans les neurones noradrénergiques. Bien qu’il y ait peu de sites DAT sur les neurones dopaminergiques de la PFC, leur capacité de recapture est suffisante pour que l’amphétamine provoque une libération substantielle de dopamine, bien qu’il ait été suggéré qu’une grande partie de la libération de dopamine dans la PFC provienne des neurones noradrénergiques.
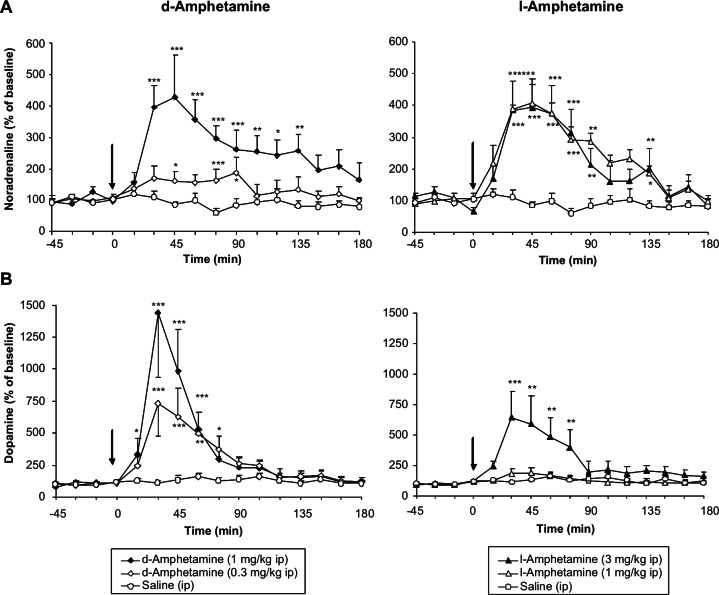
Lorsque l’on compare les profils pharmacologiques in vivo des isomères de l’amphétamine, la d-amphétamine est trois à cinq fois plus puissante que la l-amphétamine (figure 4). En outre, une analyse des effets relatifs des isomères de l’amphétamine sur les différentes catécholamines révèle que la d-amphétamine a des effets plus importants sur la dopamine que sur la noradrénaline, tandis que la l-amphétamine a une action plus équilibrée en augmentant à la fois la neurotransmission dopaminergique et la neurotransmission noradrénergique (figure 4). Bien que les effets des isomères de l’amphétamine illustrés à la figure 4 aient été obtenus chez une souche particulière de rats prédisposés à l’hypertension (SHR), des effets similaires de la d- et de la l-amphétamine ont été rapportés lors d’expériences réalisées dans la PFC et le striatum de souches non hypertendues. Kuczenski et al. (1995) ont déterminé les effets des deux énantiomères de l’amphétamine sur la libération de 5-HT dans le caudé. L’effet était considérablement plus faible que celui observé pour la dopamine et la différence de puissance entre les deux isomères était plus faible.
Plus haut dans cette revue, nous avons décrit la formulation de la MES-amphétamine. Des expériences in vivo ont également été réalisées pour explorer l’interaction entre le rapport 3:1 des isomères d et l dans cette formulation. Les expériences ont été réalisées sur des rats anesthésiés en utilisant la voltampérométrie in vivo pour déterminer la concentration extracellulaire de dopamine dans le striatum et le noyau accumbens. En utilisant cette technique, Joyce et al. (2007) ont démontré que la dynamique de la d-amphétamine sur l’efflux de dopamine n’était pas modifiée par la présence de l’isomère l dans le rapport 1:1 présent dans le racémate, mais que dans le mélange 3:1 d-isomère l, la l-amphétamine augmentait et prolongeait de manière significative l’efflux de dopamine dans le striatum du rat produit par la d-amphétamine. Les auteurs ont émis l’hypothèse que la l-amphétamine contenue dans la MES-amphétamine module l’activité de la DAT de manière à prolonger l’action de l’isomère d. Une autre explication de la prolongation observée de l’effet pharmacologique est que le rapport 3:1 entre les isomères d et l dans la formulation de la MES-amphétamine est optimisé de manière fortuite de sorte que l’entrée de l’isomère d dans les terminaux nerveux catécholaminergiques est modulée par la concurrence de l’isomère l pour la DAT, prolongeant ainsi l’action libératrice de neurotransmetteurs de l’isomère d le plus puissant.
Implications cliniques.
L’action principale de l’amphétamine est d’augmenter les concentrations synaptiques de neurotransmetteurs monoaminergiques, renforçant ainsi indirectement la neurotransmission noradrénergique et dopaminergique dans le CNS. Bien que les isomères de l’amphétamine soient également de puissants agents de libération de la 5-HT in vivo, cette action ne semble pas contribuer à leur efficacité dans le traitement du TDAH. Cette opinion est basée sur l’expérience clinique avec la fenfluramine, qui est un analogue chimique de l’amphétamine et un puissant agent libérateur ayant une action préférentielle sur la 5-HT. Donnelly et al. (1989) ont rapporté que la fenfluramine n’était pas efficace pour traiter les comportements perturbateurs et hyperactifs du TDAH ; elle n’a pas non plus amélioré le trouble des conduites présent chez environ la moitié des sujets. Cependant, il est possible que l’action de l’amphétamine sur l’augmentation de la force sérotoninergique ait un effet bénéfique sur l’anxiété ou la dépression qui sont souvent comorbides avec le TDAH. Ainsi, l’augmentation de la signalisation catécholaminergique est le principal médiateur de l’efficacité de l’amphétamine dans le TDAH et la narcolepsie. En revanche, cette même pharmacologie est également responsable des principaux effets secondaires de l’amphétamine et de son risque d’abus récréatif. Par conséquent, l’optimisation de l’efficacité thérapeutique tout en maintenant les effets secondaires à un niveau acceptable est un équilibre difficile à atteindre, qui nécessite un titrage minutieux de la dose chez le patient.
Efficacité.
Il est admis depuis longtemps que le TDAH s’accompagne d’un dérèglement des systèmes catécholaminergiques du PFC et de ses connexions avec les régions sous-corticales, y compris le striatum. Des études de neuro-imagerie chez des sujets atteints de TDAH ont révélé des altérations anatomiques et des changements fonctionnels compatibles avec une réduction de la fonction dopaminergique dans diverses régions du cerveau riches en dopamine, notamment le cortex frontal, le striatum et le globus pallidus.
Sur la base des observations selon lesquelles les isomères de l’amphétamine provoquent des augmentations très importantes et rapides de l’efflux de dopamine et de noradrénaline dans le PFC et de dopamine dans le striatum, il a été prédit que ces médicaments seraient très efficaces dans le traitement du TDAH. Cette hypothèse a été confirmée par des rapports faisant état de l’efficacité de la d-amphétamine, de la l-amphétamine, de l’amphétamine racémique et de la MES-amphétamine dans le traitement du TDAH. Il est généralement admis que l’efficacité des amphétamines n’est pas différente de celle du méthylphénidate, qui est l’autre stimulant majeur utilisé pour traiter le TDAH. Toutefois, une méta-analyse réalisée par Faraone et Buitelaar (2010) a montré que les médicaments à base d’amphétamine étaient modérément plus efficaces. Cela concorde avec les résultats précliniques selon lesquels le méthylphénidate renforce aussi nettement la pulsion catécholaminergique dans le PFC et le striatum. D’autre part, plusieurs essais ont fait état de l’efficacité supérieure de l’amphétamine dans le traitement du TDAH par rapport à l’atomoxétine (Strattera®), un inhibiteur sélectif de la recapture de la noradrénaline non stimulant. Cette constatation concorde avec les résultats d’expériences de microdialyse in vivo qui ont montré que l’atomoxétine peut produire des augmentations modérées de la noradrénaline et de la dopamine extracellulaires dans le PFC en bloquant l’entrée de ces deux neurotransmetteurs catécholaminergiques dans les neurones noradrénergiques via les sites NET, mais en tant qu’inhibiteur sélectif de la recapture de la noradrénaline, il est sans effet dans d’autres régions du cerveau, telles que le striatum et le noyau accumbens, où les concentrations synaptiques de dopamine sont régulées par les sites DAT.
Sécurité et effets indésirables.
Avec les applications cliniques de l’amphétamine comme médicament contre la fatigue, comme coupe-faim et comme traitement de la narcolepsie, les effets indésirables tels que l’anorexie, la perte de poids et l’insomnie sont des effets indésirables prévisibles et fréquents associés à l’utilisation de médicaments à base d’amphétamine dans la prise en charge du TDAH. Ces effets secondaires ont été rapportés pour la d-amphétamine, la MES-amphétamine. Parmi les autres effets indésirables provoqués par les amphétamines figurent les nausées, les vomissements, les crampes abdominales, l’augmentation de la pression artérielle et de la fréquence cardiaque et, éventuellement, l’exacerbation des tics moteurs.
Responsabilité en cas d’abus.
Les stimulants ont tendance à être appréciés par une certaine proportion de la population, mais pas par tout le monde, loin s’en faut. Il semble que le tonus dopaminergique basal détermine ce phénomène, les personnes qui ont un nombre plus élevé de récepteurs dopaminergiques D2, mesuré par tomographie par émission de positons (TEP) au [11C]-raclopride, trouvant les stimulants aversifs plutôt qu’agréables. Cependant, l’expérience agréable que procure la d-amphétamine peut conduire à une utilisation excessive du médicament prescrit par le patient et à une (mauvaise) utilisation de la prescription par d’autres personnes (détournement). Pour ces raisons, tous les traitements actuels à base de stimulants de type amphétamine sont des drogues contrôlées en vertu de la loi britannique de 1971 sur l’abus des drogues), tous les membres de cette catégorie étant placés dans la classe B, à l’exception de la méthamphétamine, qui a récemment été placée dans la classe A par crainte d’une explosion de l’abus récréatif similaire à celle observée aux États-Unis et en Thaïlande.
En réalité, les patients atteints de TDAH abusent peu de ces médicaments et, dans la plupart des cas, le défi pour le médecin prescripteur est de faire en sorte que les patients continuent à prendre leurs médicaments plutôt que d’en limiter l’usage. De nombreux patients adolescents cessent de prendre leurs médicaments malgré les avantages évidents qu’ils en retirent pour leurs résultats scolaires ; ils invoquent des raisons telles que le sentiment d’être trop contrôlés, le désir d’être responsabilisés par les médicaments, etc. Pour ces raisons, les observations de dépendance et d’abus de d-amphétamine sur ordonnance sont rares dans la pratique clinique, et ce stimulant peut même être prescrit à des personnes ayant des antécédents d’abus de drogues, à condition que certains contrôles, tels que le retrait quotidien des ordonnances, soient mis en place.
Il est bien connu que les toxicomanes récréatifs et les utilisateurs dépendants administrent généralement des psychostimulants à des doses plusieurs fois supérieures à celles stipulées pour un usage thérapeutique. En outre, pour obtenir un effet pharmacologique maximal, la quantité maximale de drogue doit être délivrée dans le CNS dans le temps le plus court possible. C’est cet impératif qui pousse les toxicomanes à passer de méthodes d’auto-administration relativement sûres, comme l’ingestion orale, à des voies de plus en plus dangereuses, comme renifler la cocaïne, la fumer (“crack” ou “crystal meth”) ou l’injecter par voie intraveineuse. Un autre facteur moins connu de l’abus de drogues est le désir de gratification instantanée des consommateurs. Ainsi, l’attrait d’une drogue particulière en tant que substance récréative est dans une large mesure déterminé par sa capacité à produire les effets désirés en quelques minutes, par exemple le “rush” de la cocaïne.
La cinétique de la d-amphétamine administrée par voie orale la rend moins gratifiante (agréable) que la cocaïne ou la méthamphétamine. La cocaïne, qu’elle soit sniffée ou fumée sous forme de crack, pénètre très rapidement dans le cerveau et semble même s’y concentrer par rapport au plasma, ce qui explique le fort potentiel de récompense de cette drogue : une pénétration plus rapide dans le cerveau entraîne un “high” plus important. La méthamphétamine pénètre plus lentement dans le cerveau et ses effets maximaux sont retardés de 10 à 15 minutes par rapport à la cocaïne. Bien que le sulfate de d-amphétamine n’ait pas été étudié de manière exactement comparable, ses propriétés physico-chimiques nous permettent de prédire qu’après ingestion orale, le taux d’absorption de la d-amphétamine dans le cerveau serait encore plus lent que celui de la méthamphétamine. Cela dit, l’abus de d-amphétamine n’est pas une raison pour se reposer sur ses lauriers. Bien que l’abus d’amphétamine ait atteint son apogée dans les années 1960, l’utilisation abusive d’amphétamine est un problème social, juridique et médical persistant. L’usage intraveineux de d-amphétamine et d’autres stimulants présente toujours des risques majeurs pour la sécurité des personnes qui s’y adonnent. Une partie de cet abus intraveineux provient du détournement d’ampoules de d-amphétamine, qui sont encore occasionnellement prescrites au Royaume-Uni pour le contrôle de la narcolepsie sévère et d’autres troubles de sédation excessive. Toutefois, la majeure partie de la d-amphétamine intraveineuse provient de la production illicite locale. Certains toxicomanes utilisent des solvants pour extraire l’ingrédient actif des comprimés ou des capsules, qui peuvent ensuite être concentrés et injectés par voie intraveineuse. Le développement de formulations de d-amphétamine inviolables a été un objectif majeur de l’industrie pharmaceutique pour prévenir ce type d’abus. Plusieurs nouveaux médicaments sur ordonnance contenant de la d-amphétamine à prise unique quotidienne sont apparus et présentent un degré élevé de dissuasion de la falsification, par exemple l’Adderall XR. En outre, la lisdexamfétamine, un promédicament de la d-amphétamine, constitue une avancée supplémentaire dans la réduction du risque de détournement, car elle permet une augmentation plus progressive de la concentration de la drogue dans le cerveau, réduisant ainsi davantage les effets agréables de la d-amphétamine. Nous reviendrons sur ces sujets dans la suite de cette étude.
Volkow et ses collègues ont effectué un nombre considérable de recherches en utilisant la TEP et d’autres techniques d’imagerie cérébrale pour explorer la relation entre l’occupation du DAT, la concentration synaptique de dopamine et l’occupation des récepteurs D2 de la dopamine pour les drogues psychostimulantes dont on abuse. Bien que l’hypothèse de la libération de dopamine dans le cadre du renforcement par la drogue, proposée par Di Chiara et Imperato (1988) sur la base d’expériences réalisées sur des rats, puis étendue à l’homme par Volkow et ses collègues, ait ses limites, il est désormais bien admis que l’euphorie, la psychostimulation et le renforcement produits par les drogues stimulantes se produisent lorsqu’il y a des augmentations rapides et substantielles des concentrations synaptiques de dopamine dans le striatum basal et dans le système mésolimbique du cerveau humain. Ces chercheurs ont également démontré que le taux d’occupation du DAT par des drogues telles que la cocaïne et le méthylphénidate est essentiel pour leur capacité à produire des “highs” chez les sujets humains. Bien que la d-amphétamine soit un substrat compétitif de la DAT plutôt qu’un inhibiteur classique de la recapture, les mêmes principes s’appliquent à son action pharmacologique. Ainsi, le taux et l’ampleur de la libération de dopamine neuronale produite par l’amphétamine dépendent absolument du taux et de la concentration de drogue qui atteint les sites DAT dans le cerveau. Peu de recherches ont été menées chez l’homme sur cette cinétique à l’aide de l’imagerie cérébrale, mais il semble probable que les mêmes règles s’appliquent.
Conformément aux résultats des expériences de microdialyse, la d-amphétamine est plus puissante que la l-amphétamine pour évoquer des effets subjectifs de type stimulant chez les rats et une activation comportementale chez les primates. Une généralisation croisée se produit entre les indices subjectifs évoqués par les isomères d et l de l’amphétamine, ce qui indique un mécanisme neurochimique commun. Il a été démontré que les deux isomères de l’amphétamine servent de renforçateurs positifs chez les animaux (c’est-à-dire que les animaux travaillent pour obtenir plus de drogue). Il en va de même chez l’homme, l’isomère d étant à nouveau deux à trois fois plus puissant que l’isomère l. En partant du principe que les effets subjectifs et renforçateurs des isomères de l’amphétamine se transposent bien de l’animal à l’homme, et en supposant que les médiateurs neurochimiques sont également cohérents d’une espèce à l’autre, nous pouvons utiliser les résultats des expériences de microdialyse pour tirer quelques conclusions sur ce sujet. Les résultats de la figure 4, qui révèlent que les deux isomères sont des libérateurs de noradrénaline aussi puissants l’un que l’autre, mais que la d-amphétamine est environ trois fois plus puissante que la l-amphétamine en tant que libérateur de dopamine, indiquent que la dopamine est le principal médiateur neurochimique des propriétés stimulantes et euphorisantes de l’amphétamine. Comme indiqué ci-dessus, c’est la combinaison d’un taux d’augmentation rapide et de l’ampleur de l’effet qui explique les puissants effets stimulants de l’amphétamine.
Bien que la l-amphétamine soit le moins puissant des deux isomères, son efficacité pharmacologique ne doit pas être sous-estimée. Cheetham et al. (2007) ont rapporté que les deux isomères étaient capables d’augmenter l’efflux de dopamine striatale de plus de 5 000 % par rapport aux valeurs de base, ces effets atteignant leur maximum en 45 minutes environ. En revanche, les augmentations maximales de l’efflux de dopamine obtenues avec les inhibiteurs classiques de la recapture de la dopamine (par exemple le bupropion et le GBR 12909) sont cinq à dix fois plus faibles et prennent souvent plus d’une heure pour atteindre leur maximum. L’importance du taux d’augmentation des concentrations synaptiques de dopamine pour l’induction de la stimulation et de l’euphorie est illustrée par l’observation que le bupropion et le GBR 12909 n’ont pas été ressentis comme stimulants ou euphorisants par des volontaires normaux ou par des consommateurs récréatifs expérimentés de stimulants. Dans les essais sur le bupropion et le GBR 12909 où la d-amphétamine était utilisée comme témoin positif, ses effets stimulants, énergisants et renforçants ont été reconnus sans équivoque par les sujets normaux et les consommateurs de drogues à usage récréatif.
Formules à prise unique quotidienne.
Dans les revues précédentes, nous avons décrit en détail l’efficacité et la sécurité des médicaments stimulants et non stimulants utilisés dans la prise en charge du TDAH et comparé les mérites relatifs de chacun d’entre eux. Cette analyse a révélé que les stimulants, y compris l’amphétamine, sont toujours considérés comme les médicaments les plus efficaces disponibles. Certaines tentatives d’introduction de nouveaux médicaments, comme la guanfacine XR (Intuniv®), ont été couronnées de succès, mais beaucoup d’autres nouvelles approches pharmacologiques ont échoué (voir Heal et al., 2012). D’autre part, les innovations en matière de technologie de formulation et de systèmes d’administration de médicaments ont permis de faire des progrès considérables dans l’amélioration de la prise en charge clinique du TDAH. Tous les stimulants ont des demi-vies biologiques qui nécessitent au moins deux prises quotidiennes pour être efficaces pendant 12 à 14 heures. Le TDAH se caractérise par l’inattention, la distractibilité, des déficits de la mémoire de travail et l’impulsivité, et les sujets atteints de ce trouble sont donc particulièrement peu enclins à se conformer à des schémas d’administration rigides. L’amphétamine présentant un risque élevé d’abus récréatif, le fait de mettre les médicaments entre les mains des enfants augmente le risque de détournement ou d’abus, tandis que l’approche alternative consistant à confier les médicaments aux autorités scolaires nécessite des installations appropriées pour le stockage des médicaments contrôlés. L’administration d’un médicament stimulant à prise unique quotidienne à un enfant ou à un adolescent dès le matin, sous la surveillance de ses parents, le dispense de prendre des médicaments supplémentaires à l’extérieur du domicile, et évite au patient de devoir prendre des médicaments supplémentaires dans des délais très stricts. L’un des autres avantages de ces nouvelles formulations est qu’elles sont inviolables et qu’il est donc difficile pour les toxicomanes d’extraire l’amphétamine pour l’auto-administrer par des voies dangereuses, telles que la fumée, le reniflement ou l’injection intraveineuse. Parmi les exemples de médicaments à base d’amphétamine à prise unique quotidienne, on peut citer le MES-Amphétamine XR et le promédicament de la d-amphétamine, la lisdexamfétamine.
Lisdexamfetamine.
Comme indiqué brièvement plus haut, la lisdexamfétamine est le premier promédicament amphétaminique dont l’utilisation a été approuvée pour le traitement du trouble déficitaire de l’attention avec hyperactivité. La lisdexamfétamine n’a pas d’affinité pour un large éventail de transporteurs, dont le DAT et le NET (Vyvanse®, étiquette du produit aux États-Unis) ou de récepteurs, de canaux ioniques, de sites de liaison allostérique et d’enzymes (tableau 3). Ce profil est cohérent avec le fait que la lisdexamfétamine est pharmacologiquement inactive. Bien qu’il n’existe pas d’informations définitives sur le sujet, la grande taille moléculaire et les caractéristiques polaires de la lisdexamfétamine indiquent qu’il est peu probable que la molécule mère traverse la barrière hémato-encéphalique. Des expériences in vitro ont révélé que le métabolisme de la lisdexamfétamine en d-amphétamine se produit dans les globules rouges par hydrolyse enzymatique à vitesse limitée.
[TABLEAU 3]
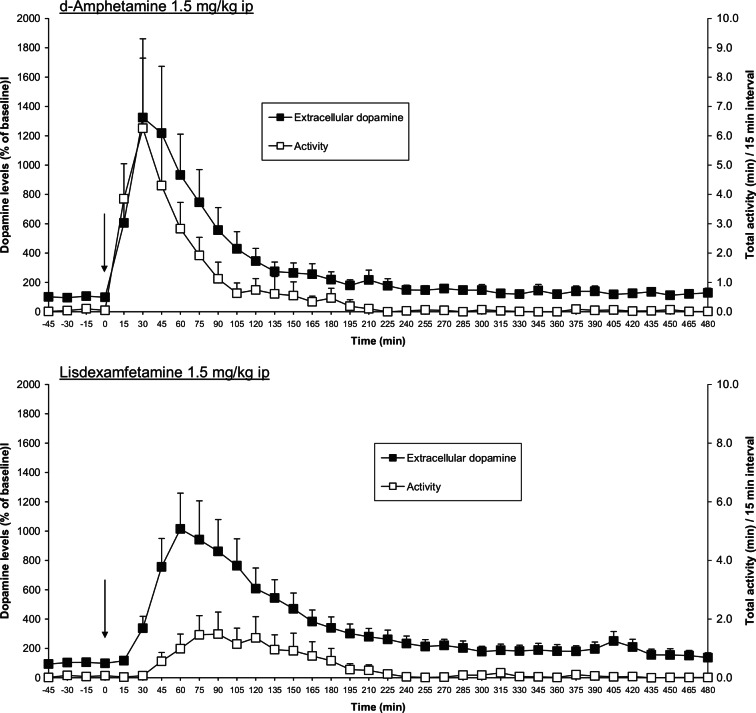
Les relations pharmacocinétique/pharmacodynamique (PK/PD) de la lisdexamfétamine et du sulfate de d-amphétamine à libération immédiate (IR) ont été étudiées chez des rats, où l’échantillonnage sanguin automatisé a été combiné à l’échantillonnage du microdialysat striatal. L’activité locomotrice des rats a également été contrôlée simultanément. Après administration de doses équivalentes de lisdexamfétamine et de d-amphétamine IR (1,5 mg/kg ip sous forme de d-amphétamine base), les profils pharmacocinétiques plasmatiques observés pour la fraction pharmacologiquement active, la d-amphétamine, étaient très différents. Les valeurs de l’AUC0-480 min étaient identiques, mais la concentration maximale atteinte dans le plasma (la Cmax) était 50 % plus faible après l’administration de lisdexamfétamine et le temps nécessaire pour atteindre la Cmax (le tmax) était doublé. Ces observations sont tout à fait cohérentes avec l’hypothèse d’une conversion enzymatique de la lisdexamfétamine en d-amphétamine à vitesse limitée. Cette différence dans les caractéristiques pharmacocinétiques a eu un impact profond sur les effets pharmacologiques de ces deux composés chez le rat (Figure 5). La lisdexamfétamine a produit une augmentation progressive et soutenue de l’efflux de dopamine striatale, alors que l’augmentation produite par la d-amphétamine IR a été plus rapide, atteignant un pic à 30 minutes, et a ensuite diminué plus rapidement (Figure 5).
Les effets de la d-amphétamine sur l’efflux striatal de dopamine et sur l’activité locomotrice sont superposables (figure 5), c’est-à-dire que la libération rapide de dopamine se traduit directement par une augmentation immédiate et substantielle de l’activité locomotrice. Dans le cas de la lisdexamfétamine, l’augmentation plus graduelle et plus soutenue de l’efflux de dopamine est associée à une réponse locomotrice beaucoup plus faible et visiblement retardée. En utilisant une analyse de l’hystérésis pour définir la relation entre les composantes ascendante et descendante d’une courbe de concentration en fonction du temps pour la concentration extracellulaire de dopamine dans le striatum et la réponse fonctionnelle (activité locomotrice), la relation était dans le sens inverse des aiguilles d’une montre pour la lisdexamfétamine, mais dans le sens des aiguilles d’une montre pour l’IR d-amphétamine (p < 0,05). En utilisant l’analyse de l’hystérésis de manière plus conventionnelle pour explorer la relation entre la concentration plasmatique de d-amphétamine et la réponse fonctionnelle, on a constaté une nette différence entre les deux composés, avec une hystérésis dans le sens inverse des aiguilles d’une montre pour la lisdexamfétamine et aucune hystérésis pour l’IR d-amphétamine. L’hystérésis antihoraire montre que l’effet fonctionnel de la lisdexamfétamine était plus important lorsque la concentration plasmatique de d-amphétamine diminuait, tandis que l’absence d’hystérésis avec la d-amphétamine IR démontre que dès que la concentration plasmatique du médicament commence à diminuer, son effet pharmacologique diminue également.
L’importance clinique de ces résultats sera discutée dans la section suivante.
Implications de la pharmacocinétique de la lisdexamfétamine pour l’efficacité, la sécurité et le risque d’abus récréatif.
L’efficacité de la lisdexamfétamine a été démontrée dans un certain nombre d’essais cliniques randomisés, en double aveugle, contrôlés par placebo, dans le traitement du TDAH chez l’enfant, l’adolescent et l’adulte. La lisdexamfétamine ayant fait l’objet de plusieurs études, nous nous concentrerons sur la contribution probable du profil pharmacocinétique et pharmacodynamique particulier de la lisdexamfétamine à son efficacité en tant que traitement du TDAH et à son potentiel d’abus récréatif et de dépendance plus faible que celui de l’amphétamine.
Biederman et al. (2007a) ont publié les résultats du seul essai clinique dans lequel l’efficacité et la sécurité de la lisdexamfétamine dans le traitement du TDAH ont été comparées directement à celles d’un autre médicament ayant fait ses preuves en clinique, le MES-amphetamine XR. Après une période de rodage ouverte de 3 semaines au cours de laquelle la dose de MES-amphetamine XR a été optimisée à 10, 20 ou 30 mg une fois par jour, les sujets ont été randomisés dans un essai croisé à 3 voies en double aveugle, contrôlé par placebo. Ils ont reçu leur dose optimale de MES-amphetamine XR, une dose équivalente de lisdexamfetamine en termes de d-amphétamine base, ou un placebo. Sur les variables d’efficacité primaires et secondaires que sont le comportement, l’attention et la résolution de problèmes, la lisdexamfétamine a eu une efficacité équivalente ou supérieure à celle de la MES-amphétamine XR, les deux médicaments ayant une efficacité maximale 2 heures après la prise (Biederman et al., 2007a). Cependant, en ce qui concerne les critères d’évaluation de la résolution de problèmes, il est également évident que la lisdexamfétamine a maintenu son effet maximal pendant au moins 12 heures, alors que l’effet de la MES-amphetamine XR a montré une nette diminution après 6-8 heures (Biederman et al., 2007a). Une durée d’effet exceptionnellement longue de la lisdexamfétamine a été observée par Wigal et al. (2009, 2010b), qui ont rapporté que des améliorations significatives du comportement et de l’attention chez des enfants souffrant de TDAH ont été observées dès 1 heure après l’administration de lisdexamfétamine, et que son efficacité sur le comportement, l’attention et la résolution de problèmes s’est maintenue jusqu’à 13 h. Une analyse post-hoc des données a également montré que le sexe et l’âge des sujets n’avaient pas d’influence significative sur l’efficacité de la lisdexamfétamine.
Ces observations correspondent bien au profil pharmacodynamique de la lisdexamfétamine dans les expériences de microdialyse. Ainsi, une dose de lisdexamfétamine n’entraînant qu’une faible augmentation de l’activité locomotrice a provoqué une augmentation de > 500 % de l’efflux de dopamine au niveau striatal, qui s’est maintenue pendant au moins 6 heures (figure 5). Des études pharmacocinétiques menées sur des sujets humains ont révélé que le tmax de la d-amphétamine plasmatique est atteint environ 3 heures après la prise de lisdexamfétamine ; par la suite, la d-amphétamine plasmatique diminue de telle sorte qu’à 12 heures, sa concentration est tombée à environ 60 % de la Cmax. Le maintien de l’effet thérapeutique dans le TDAH lorsque les concentrations plasmatiques de d-amphétamine diminuent indique que l’hystérésis antihoraire observée dans les expériences précliniques de pharmacocinétique et de pharmacodynamique s’applique probablement aussi à son efficacité clinique.
Une autre façon de produire une augmentation plus douce de la dopamine cérébrale consiste à lier la d-amphétamine à un support. MES-amphetamine XR utilise une technologie de billes pour délivrer deux doses bolus d’amphétamine, la première immédiatement et la seconde environ 4 heures plus tard, ce qui donne un Cmax pour les isomères d et l de l’amphétamine de 6 à 8 heures (Adderall XR®, étiquette du produit aux États-Unis). Par conséquent, l’effet thérapeutique maximal de la MES-Amphétamine XR coïncide presque exactement avec le tmax de la d-amphétamine plasmatique (Adderall XR®, étiquette du produit aux États-Unis). Ces résultats sont également cohérents avec la relation PK/PD préclinique pour la d-amphétamine IR qui a révélé une absence d’hystérésis entre la concentration plasmatique de d-amphétamine et la réponse fonctionnelle, c’est-à-dire l’activité locomotrice.
Un autre facteur qui contribue presque certainement au niveau élevé et constant d’efficacité thérapeutique observé avec le traitement à la lisdexamfétamine est la très faible variabilité inter- et intra-sujet de la concentration plasmatique de d-amphétamine observée après l’administration du promédicament par rapport aux stimulants formulés de manière traditionnelle, y compris les formulations à libération perlée et osmotique. Une fois encore, la pharmacocinétique reproductible de son métabolite actif, la d-amphétamine, est probablement due au clivage enzymatique à vitesse limitée de la molécule précurseur qui se produit principalement dans les globules rouges.
Dans deux études publiées précédemment, Jasinski et Krishnan ont comparé les effets subjectifs de la lisdexamfétamine et de la d-amphétamine IR chez des volontaires humains ayant déjà consommé des drogues, lorsque ces composés étaient administrés par voie intraveineuse (Jasinski et Krishnan, 2009a) et par voie orale. Dans l’essai où ils ont comparé ces composés après administration orale, la d-amphétamine IR (40 mg (29,6 mg de d-amphétamine base)) a provoqué une augmentation statistiquement significative par rapport au placebo de la “préférence pour la drogue” sur l’échelle du Drug Rating Questionnaire – Subject (DQRS), alors que la dose équivalente de lisdexamfétamine (100 mg, par voie orale) ne l’a pas fait. En outre, le pic de l’effet pharmacologique de la lisdexamfétamine a été considérablement retardé par rapport à celui de la d-amphétamine à LI, soit 3 heures contre 1,5 à 2 heures. Lorsque la lisdexamfétamine a été administrée à une dose plus élevée de 150 mg, elle a augmenté de manière significative le score DQRS “goût pour la drogue” dans une mesure équivalente à celle de la d-amphétamine à LI (40 mg par voie orale). Cependant, l’effet maximal de la dose plus élevée de lisdexamfétamine a été encore plus tardif, à 4 heures.
Lorsque la voie intraveineuse a été explorée, la d-amphétamine IR (20 mg par voie intraveineuse) a produit un pic de “goût du médicament” 20 minutes après l’administration, ce qui coïncidait avec la Cmax plasmatique. En revanche, la dose équivalente de lisdexamfétamine (50 mg par voie intraveineuse) n’a pas augmenté de manière significative le “Dug liking” par rapport au placebo, et la Cmax de la d-amphétamine plasmatique est apparue beaucoup plus tard, à 2 heures (Jasinski et Krishnan, 2009b). Les deux composés ont produit des valeurs équivalentes d’AUC0-24h, mais par rapport à la dose équivalente de d-amphétamine IR, la Cmax de la d-amphétamine plasmatique était trois fois plus petite pour la lisdexamfétamine et le tmax était trois fois plus grand. Ces différences dans les caractéristiques pharmacocinétiques et pharmacodynamiques de la d-amphétamine et de la lisdexamfétamine à LI observées chez l’homme par Jasinski et Krishnan (2009b) sont très similaires aux résultats de l’étude pharmacocinétique et pharmacodynamique chez le rat décrite plus haut dans cette revue.
Ces résultats permettent de conclure que, bien qu’en termes d’équivalents base de d-amphétamine, la lisdexamfétamine soit nettement moins puissante que la d-amphétamine IR, elle produit néanmoins des effets subjectifs similaires à ceux de la d-amphétamine chez l’homme. Il est également raisonnable de supposer que si la dose intraveineuse de lisdexamfétamine avait été augmentée, son effet “Drug liking” aurait été différent de celui du placebo. Cependant, lorsqu’on considère le potentiel d’abus récréatif d’une drogue, le temps nécessaire pour produire une réponse maximale est probablement aussi important que son ampleur. Dans le cas de l’IR d-amphétamine, son effet subjectif maximal est apparu beaucoup plus tôt que celui de la lisdexamfétamine, et le passage à la voie intraveineuse a accéléré le début de l’action de l’IR d-amphétamine et augmenté sa puissance. Bien que l’augmentation de la dose de lisdexamfétamine ait accru son efficacité, elle a aussi progressivement retardé le moment où l’effet est maximal. En outre, le passage à la voie intraveineuse pour la lisdexamfétamine semble avoir relativement peu d’influence sur le potentiel d’abus du promédicament.
Pour explorer cette possibilité plus avant, nous avons effectué une analyse post-hoc sur les données des rapports d’étude clinique originaux afin de comparer la pharmacodynamique et la pharmacocinétique de la lisdexamfétamine lorsqu’elle est administrée par la voie clinique (orale) par rapport à l’une des voies privilégiées par les abuseurs récréatifs (intraveineuse). Ce sujet est particulièrement important car la lisdexamfétamine a une solubilité aqueuse très élevée, ce qui rend le promédicament très facile à extraire. En fait, briser la gélule et dissoudre le contenu dans l’eau est indiqué comme une voie d’administration pour les patients incapables d’avaler des gélules (Vyvanse®, étiquette du produit aux États-Unis).
Comme le montre le tableau 4, les scores maximaux moyens sur les échelles DQRS et Drug Rating Questionnaire – Observer (DRQO) pour “aimer”, “ressentir l’effet du médicament” et “ne pas aimer” révèlent que les effets subjectifs de la lisdexamfétamine (50 mg) n’étaient pas significativement différents lorsque le promédicament était administré par voie orale ou par voie intraveineuse. Ce résultat montre que les effets subjectifs de la lisdexamfétamine n’ont pas été augmentés lorsque le médicament a été administré par voie intraveineuse. Les mesures de la pression artérielle sont des mesures objectives utiles des effets pharmacodynamiques des médicaments sympathomimétiques. Par rapport au placebo, 50 mg de lisdexamfétamine ont augmenté de manière significative le pic de pression artérielle systolique lors de l’administration par voie orale et intraveineuse et la pression artérielle diastolique lors de l’administration par voie orale (figure 6). Il ressort également des données de la figure 6 que l’ampleur des augmentations de la pression artérielle systolique et diastolique n’était pas statistiquement différente après l’administration orale ou intraveineuse de lisdexamfétamine.
[TABLEAU 4]
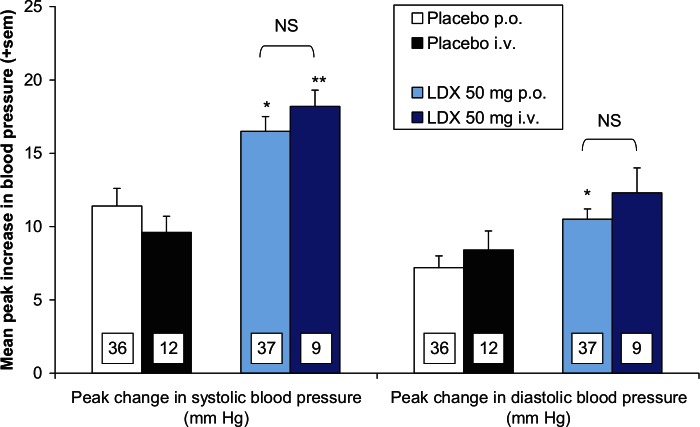
Les paramètres pharmacocinétiques de la d-amphétamine plasmatique observés après administration orale ou intraveineuse de lisdexamfétamine (50 mg) sont également résumés dans le tableau 4. L’AUC0-infinité montre que l’exposition globale au médicament est identique quelle que soit la voie d’administration. Il est important de noter que l’injection intraveineuse de lisdexamfétamine n’a pas augmenté de manière significative la Cmax de la d-amphétamine, ni réduit de manière significative son tmax. Bien que l’AUC0-1,0h ait indiqué que l’exposition précoce à la d-amphétamine était réduite après l’administration orale de lisdexamfétamine, cette différence s’explique probablement par le fait que la voie d’administration intraveineuse contourne le temps nécessaire à l’absorption du promédicament de l’intestin vers la circulation sanguine avant l’hydrolyse enzymatique par les globules rouges.
Ces résultats renforcent l’idée que le mécanisme inhabituel de conversion métabolique de la lisdexamfétamine en d-amphétamine a des implications importantes quant à son risque d’abus récréatif. Les effets subjectifs d’une dose de 50 mg de lisdexamfétamine étaient identiques lorsque le promédicament était administré par voie orale ou par injection intraveineuse, ce qui démontre que l’injection intraveineuse ne renforce pas la puissance pharmacologique de la lisdexamfétamine dans le CNS. Les augmentations de la pression artérielle systolique et diastolique après administration orale ou intraveineuse de lisdexamfétamine étaient également identiques, confirmant par des mesures physiologiques objectives et quantifiables que la voie d’injection intraveineuse ne renforçait pas son pouvoir pharmacologique. Ces conclusions sont étayées par les résultats pharmacocinétiques, qui montrent que l’ASC, la Cmax et le tmax ne sont pas influencés par la voie d’administration de la lisdexamfétamine.
Ces résultats sont complétés par ceux d’Ermer et al. (2011), qui ont rapporté que les profils pharmacocinétiques étaient identiques lorsque la lisdexamfétamine était administrée par voie intranasale ou orale, ce qui indique que les tentatives visant à augmenter son potentiel d’abus récréatif en la “sniffant” seraient également vaines. Bien que ces résultats ne démontrent pas que la lisdexamfétamine n’a aucun potentiel d’abus récréatif, ils indiquent que son attrait pour les abuseurs sera réduit par rapport à la d-amphétamine IR. Sur la base de ces données, la probabilité que la lisdexamfétamine soit largement consommée par voie intraveineuse ou nasale est très faible.
Conclusions.
Il y a maintenant un peu plus de cent ans que l’amphétamine a été découverte pour la première fois. Au cours de cette période, l’amphétamine est passée du statut de médicament largement disponible sans ordonnance pour le traitement d’un large éventail de troubles à celui de médicament contrôlé à usage très restreint qui, du moins en Europe, a pratiquement disparu des formulaires dans de nombreux pays. Les liens très clairs entre la structure moléculaire et le mode d’action pharmacologique et, à son tour, l’efficacité et la sécurité chez l’homme, font de l’amphétamine un exemple classique de validité translationnelle. La pharmacologie primaire de ces médicaments n’est pas seulement responsable de leur efficacité dans des troubles tels que le TDAH et la narcolepsie, mais aussi de leur spectre d’effets indésirables et de leur risque d’abus récréatif, ce qui fait de l’équilibre bénéfice/risque le principal défi de leur utilisation clinique. L’amphétamine se classe, avec le méthylphénidate, parmi les médicaments les plus efficaces disponibles pour la prise en charge du TDAH, et les progrès réalisés dans la mise au point de véritables médicaments à prise unique quotidienne ont permis de résoudre certains problèmes de couverture thérapeutique, tout en réduisant le risque de détournement et d’abus à des fins récréatives.
