Edinoff, A. N., Kaplan, L. A., Khan, S., Petersen, M., Sauce, E., Causey, C. D., … & Kaye, A. D. (2021). Full opioid agonists and tramadol: pharmacological and clinical considerations. Anesthesiology and Pain Medicine, 11(4).
Abstract
Les opioïdes sont des agonistes des récepteurs mu et jouent un rôle important dans le traitement de la douleur depuis des milliers d’années. Afin d’utiliser ces médicaments de manière appropriée et avec succès chez les patients, que ce soit pour contrôler la douleur, traiter les effets secondaires induits par les opiacés ou les syndromes de sevrage des opiacés, une solide compréhension de la pharmacologie de ces médicaments est cruciale. Les opioïdes agonistes complets les plus connus sont l’héroïne, la morphine, la codéine, l’oxycodone, la mépéridine et le fentanyl. Les phénanthrènes sont des composés naturels d’origine végétale qui comprennent au moins trois anneaux fusionnés. Les opioïdes dérivés de l’opium sont des dérivés du phénanthrène, alors que la plupart des opioïdes synthétiques sont des molécules plus simples qui n’ont pas d’anneaux multiples. La méthadone agit comme un analgésique opioïde synthétique similaire à la morphine, tant en qualité qu’en quantité ; cependant, la méthadone dure plus longtemps, se présente sous forme orale, est plus efficace et est considérée comme un diphénylheptane. Le fentanyl est un dérivé synthétique puissant de la phénylpipérine qui présente une activité d’agoniste opioïde mu-sélectif environ 50 à 100 fois plus puissante que la morphine. La mépéridine est un autre médicament qui est une phénylpipérine. Le tramadol est considéré comme un médicament opioïde à mécanisme mixte, car c’est un analgésique à action centrale qui exerce ses effets en se liant aux récepteurs mu et en bloquant la recapture des monoamines. Parmi les effets indésirables les plus courants de tous les opioïdes figurent les nausées, les vomissements, le prurit, la dépendance, la dépression respiratoire, la constipation, le spasme du sphincter d’Oddi et le myosis (sauf dans le cas de la mépéridine). L’utilisation chronique d’opioïdes a également établi une relation avec l’hypogonadisme induit par les opioïdes et la suppression des glandes surrénales. Les médecins doivent être les gardiens de l’utilisation des opioïdes et ne les utiliser qu’en cas de nécessité.
1. Contexte
Les opioïdes sont des agonistes des récepteurs mu et jouent un rôle important dans le traitement de la douleur depuis des milliers d’années. Afin d’utiliser ces médicaments de manière appropriée et avec succès chez les patients, que ce soit pour contrôler la douleur, traiter les effets secondaires induits par les opiacés ou le syndrome de sevrage des opiacés, il est essentiel de bien comprendre la pharmacologie de ces médicaments. Certaines caractéristiques pharmacocinétiques spécifiques, telles que la demi-vie, la clairance et le volume de distribution de ces médicaments, sont bien comprises depuis de nombreuses années. Cependant, le métabolisme et le rôle des métabolites dans la réponse pharmacodynamique chez les patients restent moins bien compris.
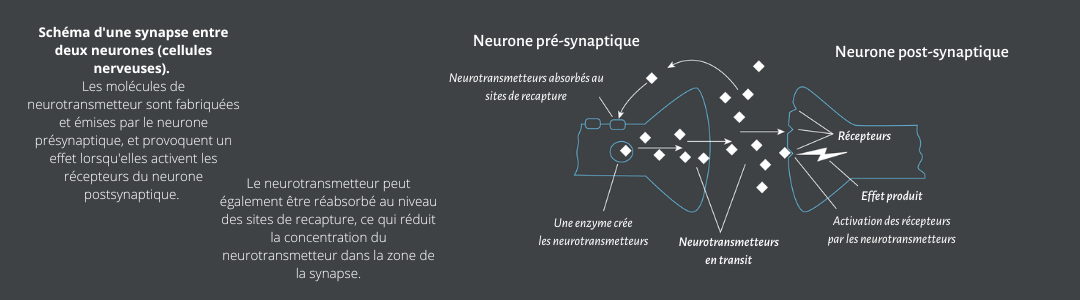
Les opioïdes activent des récepteurs transmembranaires spécifiques qui sont exprimés par les neurones centraux et périphériques, ainsi que par les cellules neuroendocrines, immunitaires et ectodermiques. Les trois principaux types de récepteurs opioïdes du système nerveux central sont mu, delta et kappa. Ces récepteurs sont classés dans le sous-groupe gamma de classe A de sept récepteurs transmembranaires couplés aux protéines G (RCPG). Les opioïdes activent ces récepteurs spécifiques qui couplent les protéines G, lesquelles initient la communication intracellulaire et activent le processus de transduction du signal. L’activation des récepteurs mu-opioïdes dans le CNS entraîne une dépression respiratoire, une analgésie, une euphorie et un myosis. Par ailleurs, la stimulation des récepteurs mu-opioïdes périphériques, tels que ceux des muscles lisses des voies gastro-intestinales et respiratoires, entraîne une suppression de la toux et une constipation induite par les opioïdes.
Les médicaments opioïdes agissent en se liant à des sites récepteurs spécifiques dans le cerveau. Ces sites sont également les sites de liaison de peptides endogènes de type opioïde qui produisent des effets similaires, y compris les effets opioïdes prototypiques de récompense, de sevrage et d’analgésie, par le biais d’actions sur ces mêmes récepteurs. On a découvert que ces trois systèmes opioïdes étaient codés par des gènes individuels pour la préproenképhaline, la préproopiomélanocortine et la préprodynorphine. Chacun de ces gènes codera pour des peptides qui se lient respectivement aux récepteurs mu (MOR), kappa (KOR) et delta (DOR). La découverte de ces peptides endogènes et de leurs récepteurs a confirmé que les médicaments opioïdes agissent en imitant ces systèmes opioïdes endogènes. MOR est la principale cible des analgésiques opioïdes, mais DOR et KOR régulent également la douleur et l’analgésie (6). Plus précisément, l’activation des récepteurs mu peut agir sur diverses protéines G qui affectent les enzymes génératrices de messagers, comme l’adénylcyclase et la phospholipase C. De manière aiguë, les opioïdes peuvent diminuer les messagers secondaires comme l’adénosine monophosphate cyclique (AMPc), tandis que l’activation chronique des récepteurs opioïdes a l’effet inverse de l’administration aiguë et entraîne une régulation à la hausse de l’AMPc.
2. Agonistes opioïdes complets – Phénanthrènes
Les phénanthrènes sont des composés naturels d’origine végétale qui comprennent trois anneaux fusionnés ou plus. Les opioïdes dérivés de l’opium sont des dérivés de phénanthrène, alors que la plupart des opioïdes synthétiques sont des molécules plus simples qui n’ont pas d’anneaux multiples. Les dérivés phénanthrènes prototypiques qui agissent comme agonistes complets du récepteur mu comprennent la morphine, l’hydromorphone et l’oxymorphone. L’héroïne (diamorphine, diacétylmorphine) est un agoniste puissant. La codéine, la dihydrocodéine, l’hydrocodone et l’oxycodone sont des agonistes légers à modérés. Certains phénanthrènes ont une action mixte sur les récepteurs et il convient d’être très prudent lors de la prescription de ces médicaments avec des agonistes purs en raison de l’imprévisibilité des effets analgésiques et de la précipitation du syndrome d’abstinence explosive. La nalbuphine est un agoniste puissant des récepteurs K et un antagoniste des récepteurs Mu, qui provoque une dépression respiratoire à des doses élevées qui n’est pas inversée par la naloxone. La buprénorphine est un dérivé du phénanthrène à longue durée d’action, agoniste partiel des récepteurs Mu et antagoniste des récepteurs K. Elle est approuvée par la FDA pour le traitement de la dépendance aux opiacés. Contrairement à la méthadone, l’administration de doses élevées de buprénorphine entraîne une action antagoniste des récepteurs Mu. Le Suboxone est une combinaison de buprénorphine et de naloxone, un antagoniste des récepteurs Mu, pour prévenir l’usage illicite par voie intraveineuse. La sédation est plus fréquente avec les opioïdes étroitement liés à la molécule de phénanthrène, alors que les agents synthétiques, tels que la mépéridine et le fentanyl, ont moins d’effets sédatifs.
Un phénanthrène, le chlorhydrate d’halofantrine, est efficace contre les stades érythrocytaires (mais pas les autres) des quatre espèces de paludisme humain. Bien qu’il ait été approuvé par la FDA, il n’est pas disponible aux États-Unis, mais l’est largement dans les pays où le paludisme est endémique.
3. Agonistes opioïdes complets – Diphénylheptanes
À la fin du XIXe siècle, les dérivés de l’opium, dont le laudanum et la morphine, étaient largement utilisés pour traiter les douleurs chroniques, au point que différentes populations, dont les soldats de la guerre civile, les médecins et les femmes au foyer, en sont devenues dépendantes. La loi Harrison de 1914 a été adoptée pour interdire le traitement d’entretien de la dépendance aux opiacés et a conduit à une augmentation massive de la consommation d’héroïne dans les rues. En 1920, la dépendance était reconnue, mais ce n’est que dans les années 1960 que la communauté scientifique a commencé à prendre conscience de la nécessité d’un programme d’entretien pour les personnes dépendantes. Pendant la Seconde Guerre mondiale, les forces alliées à l’Est contrôlaient l’approvisionnement en morphine. Des scientifiques allemands ont donc commencé à tenter de synthétiser un composé similaire, ce qui a donné naissance à la méthadone. En 1947, la méthadone est connue sous le nom de dolophine (ou dollies dans les rues), du mot dolor (douleur) et fin (fin). Dans les années 1960, Dole et Nyswander ont publié une étude à l’Institut Rockefeller préconisant l’utilisation de la méthadone pour traiter le sevrage de l’héroïne, ce qui a entraîné le remplacement de la morphine par la méthadone pour le traitement de la dépendance à l’héroïne.
La méthadone agit comme un analgésique opioïde synthétique similaire à la morphine en termes de qualité et de quantité ; cependant, la méthadone dure plus longtemps et, sous forme orale, elle est plus efficace. La méthadone est synthétisée par alkylation du diphénylacétonitrile par l’amide de sodium et le chlorure de 1-diméthylamino-2-propyle. Le résultat est combiné avec du bromure d’éthylmagnésium et ensuite hydrolysé en un mélange racémique de méthadone ou de 4,4-diphényl-6-diméthylaminoheptan3-one. En raison de la présence d’un atome de carbone asymétrique, la méthadone a deux formes énantiomériques, les isomères d et l. L’isomère l est le composant qui a le plus d’influence sur la production de méthadone. L’isomère l est le composant qui a des effets analgésiques et on a constaté qu’il avait une puissance analgésique deux fois supérieure à celle de la morphine. Alors que l’on pensait qu’il était totalement inactif, des études récentes ont montré que l’isomère d avait une activité antagoniste sur les récepteurs NMDA et pouvait jouer un rôle dans la tolérance à la morphine. Cette activité sur les récepteurs NMDA rend la méthadone plus efficace dans le traitement des douleurs neuropathiques que les autres opioïdes. La méthadone n’est disponible aux États-Unis que sous la forme d’un mélange racémique des deux énantiomères.
La méthadone est lipophile et absorbée rapidement après administration orale, avec une biodisponibilité orale de 60 à 80%. La demi-vie plasmatique est d’environ vingt-quatre heures mais varie de 5 à 130 heures, ce qui explique pourquoi il est difficile de prédire les pics plasmatiques et explique la grande variabilité de la pharmacocinétique entre les patients. La méthadone peut être détectée dans le sang environ 15 à 45 minutes après l’administration orale. La méthadone est liée aux protéines plasmatiques telles que l’α1-glycoprotéine acide, qui augmentent dans des conditions de stress telles que la dépendance à l’héroïne ; il y a donc une augmentation de la méthadone liée aux protéines et une diminution de la méthadone libre. En outre, la méthadone est également liée et distribuée dans le foie, les poumons et le tissu adipeux, ce qui explique sa longue demi-vie plasmatique. La méthadone met plusieurs jours à produire son plein effet en raison de la saturation des tissus, ce qui explique pourquoi certains usagers ont souvent l’impression de ne pas avoir assez de méthadone à bord au cours des premiers jours de traitement. Des études ont montré que les patients sous méthadone sont généralement maintenus à 80-120 mg de méthadone orale par jour, ce qui est considéré comme une dose élevée, et à 20-60 mg, ce qui est considéré comme une dose faible. Les patients commencent généralement par une dose initiale de 10 à 30 mg, qui peut être augmentée progressivement au cours des trois premières semaines. La méthadone est principalement métabolisée par le foie par déméthylation par le CYP3A4 ainsi que par de nombreux autres cytochromes. Il existe une grande variabilité en termes de propriétés pharmacocinétiques, ce qui a rendu difficile l’élaboration de recommandations en matière de dosage.
En raison de son métabolisme important par le système des cytochromes et de la durée globale du traitement d’entretien à la méthadone, la méthadone présente un risque d’interaction avec d’autres médicaments. Il a été démontré que les antidépresseurs, les anticonvulsivants, les benzodiazépines, les macrolides, les antifongiques et les antirétroviraux interagissent avec la méthadone. Il est important de noter que les inducteurs métaboliques du CYP3A4 entraînent une augmentation de l’activité enzymatique et une diminution de la quantité de méthadone facilement disponible. Cela peut entraîner des symptômes de sevrage et il est donc impératif d’observer attentivement le patient afin de garantir un dosage adéquat. Il a été démontré que la consommation chronique d’alcool entraîne une augmentation du système CYP3A4, ce qui diminue la quantité de méthadone libre disponible. Jusqu’à présent, la relation entre la dose, le niveau plasmatique et l’effet n’a pas été entièrement comprise et doit faire l’objet d’une étude plus approfondie.
3.1 Utilisation clinique de la méthadone
En 2007, les États-Unis ont perdu 55,7 milliards de dollars en raison de l’abus d’opioïdes sur ordonnance. La méthadone est un choix efficace pour maintenir les personnes dans un traitement à long terme de la dépendance aux opiacés. Le traitement d’entretien à la méthadone est associé à une augmentation globale de la productivité des patients et à une diminution du risque de rechute en raison de la capacité de la méthadone à bloquer les effets euphorisants de l’héroïne. Sa longue demi-vie permet de réduire les symptômes de sevrage et, par rapport à d’autres agonistes opioïdes, elle produit moins d’euphorie et donc moins de renforcement. La recherche a montré que le traitement d’entretien à la méthadone est également associé à une diminution de l’activité criminelle et à la capacité des toxicomanes à passer de l’usage intraveineux à l’usage parental, réduisant ainsi le risque d’infection par le VIH. Le traitement à la méthadone a en fait augmenté avec la découverte du VIH dans le but de prévenir la propagation du SIDA. La méthadone s’est également avérée efficace pour réduire les douleurs chroniques telles que les douleurs cancéreuses.
Il est particulièrement intéressant de noter que des doses de 60 à 120 mg de méthadone par jour sont associées à des taux de réussite plus élevés en raison du fait que l’héroïne est aujourd’hui plus pure que par le passé. La méthadone diffère des autres agonistes opioïdes à plusieurs égards. L’action de la méthadone est plus prolongée et l’apparition des symptômes de sevrage est moins intense mais plus longue que celle des autres. La méthadone freine aussi efficacement les effets euphorisants d’autres drogues comme la morphine et aide à stopper l’état de manque afin d’éviter les rechutes.
3.2 Effets secondaires de la méthadone
La méthadone agit principalement par l’intermédiaire du récepteur mu et est donc responsable de l’euphorie et des effets analgésiques de la méthadone ; en outre, les agonistes du récepteur mu sont associés à la constipation et à la dépression respiratoire. Des études ont montré que, par rapport à la buprénorphine, la méthadone est plus susceptible de provoquer une dépression respiratoire. La méthadone s’est avérée être un analgésique deux fois plus puissant que la morphine ; il est donc important de surveiller les patients n’ayant jamais consommé d’opiacés pour détecter les risques de dépression respiratoire et de surdosage. L’arrêt progressif de la méthadone doit se faire lentement pour éviter les symptômes de sevrage tels que l’insomnie, les nausées, les changements d’humeur, la diaphorèse et les crampes musculaires. Les risques liés à l’utilisation de la méthadone comprennent l’allongement de l’intervalle QT. Ce phénomène peut entraîner un risque d’arythmie mortelle appelée torsades de pointes. Il convient d’être prudent lorsque la méthadone est utilisée avec d’autres médicaments susceptibles d’allonger l’intervalle QT. Il peut s’agir d’antibiotiques macrolides, d’antihistaminiques non sédatifs, de certains antipsychotiques et de certains antidépresseurs.
4. Agonistes opioïdes complets – Phénylpiperdines
4.1 Le fentanyl
Le fentanyl est un dérivé synthétique puissant de la phénylpipérine qui présente une activité d’agoniste opioïde mu-sélectif, environ 50 à 100 fois plus puissant que la morphine. C’est l’un des analgésiques opioïdes les plus couramment utilisés dans l’anesthésie moderne et le traitement de la douleur. Le fentanyl a connu un décollage relativement lent après son approbation par la FDA et sa popularité n’a augmenté que précipitamment après l’introduction du concept d’anesthésie à forte dose d’opioïdes pour les patients gravement malades subissant une chirurgie à cœur ouvert et une chirurgie vasculaire. Après que les essais d’utilisation de la morphine pour cette technique se soient soldés par des patients présentant un profil d’effets indésirables important, le fentanyl/oxygène à forte dose a été étudié et s’est avéré supérieur à la morphine. Plusieurs autres voies d’administration du médicament continuent d’être étudiées et, pour l’instant, le fentanyl reste l’un des médicaments les plus populaires dans les périodes péri-opératoires.
4.2 Analogues du fentanyl : Étude structure-activité-relation
Les opioïdes peuvent être classés en groupes en fonction de leur structure chimique. Le fentanyl est le prototype de la 4-anilodopipérine, une classe très puissante d’analgésiques opioïdes synthétiques. Aujourd’hui, les relations structure-activité (SAR) sont le principal moteur de la découverte et de l’optimisation des médicaments. La substitution des analogues du fentanyl par des groupes méthyles en position trois de l’anneau pipéridine a fortement diminué la puissance du fentanyl. Ce changement suggère que la puissance de la classe des 4-anilodopipéridines est plus fortement liée aux facteurs stériques de l’encombrement et de l’isomérie cis/trans qu’à la polarité ou à la réactivité chimique. En outre, les effets de la substitution en trois positions sur la neurotoxicité sont parallèles à ceux de la puissance et de la durée d’action, ce qui suggère qu’il pourrait y avoir des récepteurs similaires responsables des effets anti-nociceptifs et neurotoxiques des opioïdes.
4.3 Formulations de fentanyl dans le traitement de la douleur : mise à jour
Initialement destiné à l’anesthésie peropératoire, le fentanyl est de plus en plus polyvalent en termes d’utilisation et de voie d’administration. Le fentanyl possède une lipophilie élevée, essentielle à son action rapide et à sa puissance. Les formulations parentérales du médicament présentaient des limites quant au lieu d’administration et un risque plus élevé de complications, ce qui a incité à développer des formulations non parentérales. La première de ces formulations a été un patch transdermique qui s’est avéré efficace pour obtenir une analgésie adéquate et régulière chez les patients atteints de cancer. L’administration buccale/transmucosale, bien que moins fréquemment utilisée, s’est avérée supérieure à la morphine à libération immédiate pour les douleurs transitoires chez les patients cancéreux. Le fentanyl inhalé par voie transpulmonaire continue d’être étudié comme une nouvelle voie d’administration potentielle. Dans l’ensemble, ces nouvelles formulations ont élargi la polyvalence du fentanyl de la période péri-opératoire à des indications dans la prise en charge de la douleur chronique et des percées douloureuses dans de multiples contextes de soins.
4.4 La mépéridine intrathécale
La mépéridine est un autre médicament de la famille des phénylpipérines. La mépéridine intrathécale est capable de fournir une analgésie et un bloc de conduction nerveuse au niveau de la racine dorsale proximale de manière irréversible par rapport à l’administration de naloxone. Elle procure une analgésie pendant environ six heures et se propage peu au niveau rostral par rapport à la morphine intrathécale (36). Par conséquent, la dépression respiratoire retardée est une complication plus rare avec la mépéridine intrathécale qu’avec la morphine. A des doses plus élevées, la mépéridine intrathécale est associée à des réactions telles que prurit sévère, nausées et sédation. En outre, des cas d’anaphylaxie ont été rapportés avec la mépéridine.
5. Utilisation des agonistes opioïdes
Les principales applications des agonistes opioïdes sont l’analgésie et l’anesthésie. Les traitements aigus par opioïdes sont considérés comme appropriés pour la prise en charge de la douleur chirurgicale et traumatique aiguë, ainsi que pour la douleur liée au cancer et la douleur non cancéreuse qui n’a pas pu être prise en charge par les non-opioïdes. Dans le contexte chirurgical, l’application directe de morphine sur les structures rachidiennes peut produire une anesthésie chirurgicale locale tout en minimisant les effets secondaires systémiques. Chez les patients souffrant de douleurs chroniques, les spécialistes du traitement de la douleur peuvent choisir d’implanter un cathéter pour l’administration programmée d’opioïdes dans l’espace épidural. Dans le cadre d’une douleur cancéreuse persistante et ininterrompue, les patchs transdermiques de fentanyl sont efficaces, tandis que les pastilles de citrate de fentanyl sont efficaces pour les percées de douleur cancéreuse et l’administration de fentanyl par la muqueuse buccale via les patchs transdermiques et les pastilles.
Les agonistes opioïdes sont uniques dans leur capacité à traiter simultanément les composantes émotionnelles et physiques de la douleur. Les agonistes opioïdes complets ou partiels présentent une activité principalement au niveau du récepteur opioïde mu (), avec une activité variable au niveau des récepteurs delta (δ) et kappa (κ) du sous-type opioïde-1 (OR-1), imitant l’activité des endorphines et enképhalines endogènes pour produire une analgésie en activant les récepteurs inhibiteurs couplés à la protéine G qui modulent la neurotransmission principalement dans le cerveau, la moelle épinière, et également dans certains sites de la périphérie. Dans le cerveau, les opioïdes se lient aux neurones qui médient la nociception dans le locus coeruleus, la moelle ventrale rostrale et la matière grise périaqueducale.
La crise actuelle des opioïdes oblige les médecins à examiner attentivement les contextes dans lesquels des analgésiques opioïdes spécifiques répondront aux besoins cliniques de patients spécifiques (39). Les opioïdes ne sont généralement pas utiles dans le traitement de la douleur nociceptive, de la douleur neuropathique, de la douleur fonctionnelle, de la douleur migraineuse ou de la douleur généralisée des tissus mous. Par conséquent, le premier rôle du médecin est d’informer le patient sur la question de savoir si les opioïdes sont un moyen efficace d’atteindre ses objectifs de gestion de la douleur pour un problème douloureux spécifique.
Le dosage efficace des médicaments opioïdes pour l’analgésie dépend en partie de variables psychologiques, notamment l’anxiété de base, la sensibilité à la douleur et la chronicité de la douleur. D’autres sources somatiques de variabilité comprennent l’exposition à la nicotine, la fonction pulmonaire et la fonction hépato-rénale. L’interaction entre ces variables nécessite des pratiques de dosage circonspectes.
La gestion des attentes est importante dans le cadre d’un traitement analgésique par opioïdes. Le médecin doit communiquer l’objectif de la thérapie opioïde ambulatoire comme étant la réduction de la douleur et l’amélioration relative de la fonctionnalité, par opposition à l’absence totale de douleur. Il est également important d’informer les patients que ceux qui répondent mal à des doses faibles ou moyennes sont mieux servis par des mécanismes alternatifs d’analgésie que par des doses plus élevées d’opioïdes.
Outre leurs effets analgésiques, les opioïdes exercent une multitude d’effets systémiques. Si nombre de ces effets sont considérés comme indésirables et recommandent des applications locales dans la mesure du possible, certains d’entre eux sont exploités pour obtenir des avantages cliniques. Il s’agit notamment des effets suivants
(1) Suppression de la toux : Les agonistes opioïdes sont des suppresseurs du réflexe de la toux à action centrale. La codéine est l’opiacé de choix pour la suppression de la toux pathologique chronique en raison de son profil favorable d’effets secondaires systémiques. Cependant, la suppression chronique de la toux crée un risque d’accumulation des sécrétions, entraînant une atélectasie ou une obstruction des voies respiratoires.
(2) Antidiarrhéique : Les agonistes opioïdes sont généralement efficaces pour contrôler la diarrhée qui n’est pas associée à une infection. Les opioïdes synthétiques comme le lopéramide et le diphénoxylate sont désormais préférés pour les applications anti-diarrhéiques en raison de leurs effets minimes sur le système nerveux central.
(3) Ischémie myocardique avec œdème pulmonaire : Pour les patients présentant une ischémie myocardique douloureuse accompagnée d’un œdème pulmonaire, la morphine est un choix courant du médecin pour la prise en charge de l’infarctus du myocarde avec sus-décalage du segment ST en l’absence de dépression respiratoire comorbide. Des modèles murins in vivo ont démontré la capacité de la morphine à réduire les lésions de reperfusion en réduisant la demande globale en oxygène du tissu cardiaque. Les données sont actuellement insuffisantes pour déterminer si ces résultats sont applicables à l’homme. Toutefois, une méta-analyse de 2019 portant sur des essais contrôlés randomisés a démontré que l’utilisation de la morphine dans les syndromes coronariens aigus entraînait une augmentation de la mortalité à l’hôpital. Les auteurs de la méta-analyse ont exprimé une grande confiance dans le fait que la morphine diminue l’effet antiplaquettaire des inhibiteurs de P2Y12, ce qui suggère un mécanisme potentiel pour l’augmentation observée de la mortalité.
(4) Atténuation des frissons induits par l’amphotéricine B : L’amphotéricine B est une thérapie antifongique administrée par perfusion qui est parfois nécessaire dans le traitement des infections fongiques opportunistes inoculées aux patients cancéreux immunodéprimés. L’administration d’amphotéricine B s’accompagne souvent de frissons, et ces effets secondaires nuisent considérablement à la qualité de vie de ces patients. L’administration d’un opioïde, la mépéridine, s’est avérée efficace pour atténuer ces effets secondaires. Bien que tous les opioïdes aient une certaine propension à atténuer les frissons, cette propriété est plus prononcée dans le cas de la mépéridine.
6. Opioïdes atypiques
Le tramadol est considéré comme un médicament opioïde à mécanisme mixte, car c’est un analgésique à action centrale qui exerce ses effets en se liant aux récepteurs mu et en bloquant la recapture des monoamines (sérotonine et norépinéphrine). Le tramadol, ainsi que son métabolite actif (M1), inhibe les voies ascendantes de la douleur en se liant aux récepteurs opiacés mu du système nerveux central, ce qui entraîne une modification de la perception de la douleur et de la réponse à celle-ci. Le tramadol et le M1 inhibent également la recapture de la sérotonine et de la noradrénaline, deux composants de la voie descendante inhibitrice de la douleur responsable du soulagement de la douleur.
Le mécanisme inhibiteur unique du tramadol, qui bloque la recapture des monoamines, comme la sérotonine et la noradrénaline, entraîne un risque d’effets secondaires associés à une disponibilité accrue des monoamines. Deux de ces effets secondaires sont le syndrome sérotoninergique et les crises d’épilepsie. Ils ne sont pas très répandus dans la population générale, mais peuvent être extrêmement dangereux s’ils ne sont pas reconnus ou traités. Le risque de syndrome sérotoninergique et de convulsions est plus élevé chez les patients présentant des comorbidités médicales, l’utilisation ou l’abus de doses supra-thérapeutiques de tramadol ou l’administration simultanée d’inhibiteurs pro-convulsivants du cytochrome P-450 sérotoninergique. Les patients qui métabolisent rapidement le cytochrome P-450 2D6 ont une réponse opioïde plus forte au tramadol et courent un risque accru d’abus ou de surdosage avec le tramadol. Les cliniciens sont donc encouragés à envisager l’utilisation de tests pharmacogénétiques pour prédire la réponse d’un individu, le risque de dépendance et donc le risque de développer un syndrome sérotoninergique ou des crises d’épilepsie. Le syndrome sérotoninergique et les crises d’épilepsie résultant de l’administration de tramadol peuvent tous deux être traités efficacement par des benzodiazépines, des soins de soutien et l’arrêt du tramadol et des autres agents responsables.
Les caractéristiques pharmacologiques particulières du tramadol lui confèrent non seulement des effets indésirables particuliers, mais aussi des avantages uniques. Le tramadol ayant une action plus douce sur les récepteurs opioïdes que les médicaments opioïdes classiques, les effets secondaires de l’administration du tramadol sont également moins importants que ceux des opioïdes classiques. Cela signifie que l’administration de tramadol présente un risque moins important de dépression respiratoire et de constipation, ainsi qu’un risque plus faible de tolérance et de dépendance au médicament. En outre, l’efficacité analgésique du tramadol a été largement prouvée dans des modèles animaux dans le contexte de la douleur aiguë et chronique.
Pendant de nombreuses années, le tramadol a été utilisé comme une alternative bien tolérée à d’autres médicaments utilisés pour le soulagement de la douleur modérée, en raison de ses activités opioïdes et monoaminergiques à “mécanisme mixte”. Cependant, ces dernières années, des études ont montré que d’autres mécanismes pouvaient être impliqués dans son activité et qu’il pouvait être utilisé de différentes manières dans la gestion de la douleur. Le tramadol a la capacité de moduler divers médiateurs impliqués dans la voie de signalisation de la douleur, ainsi que de modifier la communication entre les cellules neuronales et non neuronales à différents endroits. Ainsi, le tramadol a le potentiel de moduler l’hyperexcitabilité neuronale périphérique et centrale. Étant donné que le médicament possède un éventail aussi large de cibles moléculaires, il est en mesure de contribuer au soulagement de la douleur de nombreuses façons. Le tramadol peut être utilisé pour cibler la douleur post-opératoire, la douleur lombaire et neuropathique, ainsi que la douleur associée à l’accouchement, à l’arthrose, à la fibromyalgie et au cancer. Le tramadol constitue généralement une alternative plus puissante et souvent plus sûre au traitement de la douleur par de fortes doses d’AINS ou de faibles doses d’opioïdes plus puissants. En raison de ses effets monoaminergiques, le tramadol est également capable d’exercer des activités anxiolytiques, antidépressives et anti-frissons qui peuvent potentiellement améliorer les résultats de la prise en charge de la douleur.
7. Préoccupations relatives aux agonistes opioïdes complets
7.1 Effets secondaires individuels
Les opioïdes agonistes complets des récepteurs mu comptent parmi les médicaments les plus prescrits en raison de leur extrême efficacité en tant qu’analgésiques, mais ils s’accompagnent souvent d’effets secondaires courants. Les opioïdes agonistes complets les plus connus sont l’héroïne, la morphine, la codéine, l’oxycodone, la mépéridine et le fentanyl. La liste est longue et s’allonge, car de nouveaux métabolites sont fréquemment synthétisés pour échapper aux forces de l’ordre et pour améliorer la puissance et l’efficacité. Parmi les effets indésirables les plus communs à tous les opioïdes figurent les nausées, les vomissements, le prurit, la dépendance, la dépression respiratoire, la constipation, le spasme du sphincter d’Oddi et le myosis (sauf dans le cas de la mépéridine). Les agonistes opioïdes complets ont tendance à avoir des effets secondaires plus graves, et une utilisation abusive de ces agonistes peut entraîner le coma et la mort par dépression respiratoire.
La mauvaise gestion des opioïdes entraîne souvent une utilisation à long terme des opioïdes dans les cas de douleur aiguë, ce qui s’accompagne de ses propres effets secondaires. Des études précliniques ont suggéré que l’utilisation chronique de morphine peut supprimer les réponses du système immunitaire, et des études d’observation ont montré une association entre l’utilisation d’opioïdes à long terme et le risque d’infection. Cependant, les essais cliniques restent incohérents et le lien de causalité direct n’a pas encore été établi. L’utilisation chronique d’opioïdes a également établi une relation claire avec l’hypogonadisme induit par les opioïdes et la suppression des glandes surrénales. On a signalé que les utilisateurs d’opioïdes à long terme obtenaient de moins bons résultats lors des interventions chirurgicales parce qu’ils étaient enclins à se blesser, et l’une des théories proposées est que la suppression surrénalienne émoussait la réponse habituelle au stress lors d’une maladie aiguë. Dans le contexte chirurgical, l’utilisation chronique d’opioïdes avant l’opération est associée à une augmentation des complications postopératoires, y compris l’infection, à de moins bons résultats après l’opération, à des séjours hospitaliers plus longs et à des coûts de soins de santé plus élevés. Enfin, l’utilisation chronique d’opioïdes conduit également les individus à déclarer une douleur plus forte à l’arrêt des opioïdes. La consommation chronique d’opioïdes n’a pas seulement un impact sur le patient, car le public doit souffrir de l’augmentation des coûts des soins de santé et de l’effet que la consommation d’opioïdes prescrits et illicites a sur la société.
7.2 Effets secondaires sociaux
Les opioïdes sont connus pour être très dangereux, avec un potentiel d’abus élevé et des effets indésirables graves, y compris le coma et la mort. Au cours des cinq dernières décennies, on a assisté à une augmentation spectaculaire de la consommation d’opioïdes dans les pays développés, qui a culminé entre 2011 et 2013, et qui reste élevée. L’augmentation de la prescription et de la consommation d’opioïdes s’est accompagnée d’une hausse de la mortalité et des complications liées à la consommation d’opioïdes. Le nombre de décès dus aux opioïdes a presque triplé au cours des deux dernières décennies. Les prescriptions d’opioïdes ont conduit à des dépendances aux opioïdes, qui sont satisfaites par l’usage illicite d’opioïdes qui peut conduire à la mort. L’héroïne de rue est souvent mélangée à d’autres métabolites d’opioïdes, parfois inconnus des utilisateurs, la drogue la plus connue étant le fentanyl. Le fentanyl illicite et ses analogues constituent une menace extrêmement dangereuse pour la santé publique, car le contact avec des doses minuscules peut entraîner une exposition mortelle. Les gens consomment, sans le savoir et sans le vouloir, des produits mélangés qui conduisent à l’intoxication par les opioïdes et à la mort.
L’usage des opioïdes cible particulièrement les minorités. Alors que le taux global de délivrance d’opioïdes en 2019 était de 46,7 ordonnances pour 100 personnes, certains comtés affichaient des taux six fois plus élevés ; et dans 5 % des comtés américains, suffisamment d’ordonnances d’opioïdes ont été délivrées pour que chaque personne en ait une. Les problèmes liés aux opioïdes touchent certaines régions des États-Unis plus que d’autres, en particulier les régions où vivent des minorités. Les décès dus aux opiacés touchent de manière disproportionnée les communautés noires et hispaniques. Le taux d’augmentation des overdoses d’opioïdes chez les Noirs a augmenté de 40 % entre 2015 et 2016, alors que la population globale a augmenté de 21 %. En 2017, le CDC YRBS a indiqué que les jeunes hispaniques du secondaire avaient la prévalence la plus élevée de consommation de drogues illicites sélectionnées et de mésusage d’opioïdes sur ordonnance par rapport à l’ensemble de la population des jeunes du secondaire et aux autres races/ethnies.
8. Études cliniques : Sécurité et efficacité
La morphine reste la norme en matière d’analgésie à laquelle tous les analgésiques puissants sont comparés. La pharmacocinétique et le métabolisme des divers opiacés naturels et synthétiques sont variés, ce qui confère à chaque médicament un large éventail de propriétés qui le rendent plus ou moins approprié pour des applications spécifiques. Depuis que les médecins et le grand public ont pris conscience de l’épidémie généralisée d’abus d’opioïdes aux États-Unis, la prescription de plus en plus circonspecte de ces médicaments suscite l’intérêt d’obtenir une anesthésie ou une analgésie avec une utilisation minimale de médicaments opioïdes.
Les patients traités à la morphine dont la réponse analgésique diminue peuvent souvent présenter une diminution similaire de l’analgésie et des effets secondaires systémiques, le “phénomène de tolérance croisée”. Cependant, l’expérience clinique a montré que, comme cette tolérance croisée est souvent incomplète entre les agonistes des récepteurs mu, le passage d’un patient à un autre agoniste opioïde permet d’obtenir une amélioration cliniquement significative de l’analgésie. Il s’agit d’une alternative supérieure à l’augmentation progressive des doses d’opioïdes pour compenser la tolérance.
8.1 Effets de la consommation d’opioïdes par la mère sur le nouveau-né
Bien que les médecins américains soient conscients des syndromes de sevrage néonatal depuis les années 1870, ils continuent de prescrire des opioïdes pour les douleurs courantes liées à la grossesse, notamment les douleurs lombaires, les douleurs pelviennes, les myalgies et les migraines. Dans les années 1960, l’avènement de la thérapie d’entretien à la méthadone chez les femmes enceintes dépendantes à l’héroïne a entraîné une diminution de la mortalité fœtale et une augmentation du poids à la naissance. Toutefois, il a été noté que les fœtus exposés à la thérapie d’entretien à la méthadone présentaient des symptômes de sevrage plus prononcés que les fœtus exposés uniquement à l’héroïne. Le traitement d’entretien maternel à la buprénorphine, d’apparition relativement récente, utilise un agoniste mu partiel à la place d’un agoniste mu complet (par exemple, la méthadone) pour lier les récepteurs mu avec une activité réduite mais une affinité accrue. En outre, alors que la méthadone est généralement prescrite dans le cadre d’un dosage clinique observé, la buprénorphine peut être prise par le patient ambulatoire à domicile. Il convient de noter que les fœtus nés de mères suivant un traitement d’entretien à la méthadone ou à la buprénorphine continuent d’avoir un poids de naissance significativement plus faible, se situant dans le dixième des courbes de croissance standard de la population. Bien que de nombreuses études aient examiné la possibilité d’une augmentation des rapports de cotes de malformations congénitales spécifiques chez ces enfants, et qu’il existe des preuves d’une incidence accrue de malformations cardiaques congénitales, d’anomalies du tube neural et de pieds bots, ces études n’ont pas eu la puissance statistique requise pour conclure que le petit nombre de malformations congénitales observées était lié de manière concluante à l’exposition aux opioïdes.
8.2 Examen de la prévalence de l’hypogonadisme induit par les opiacés chez les hommes
De petites études antérieures ont suggéré que l’utilisation chronique d’opiacés chez les hommes conduisait au développement d’un hypogonadisme chez jusqu’à 90 % des patients en raison d’une augmentation de la sécrétion de testostérone et d’une suppression de l’axe HPA. Cependant, une étude de cohorte rétrospective réalisée en 2019 sur 53 888 hommes ayant pris des opioïdes pendant une période de 90 jours ou plus a été comparée à des témoins ayant pris des opioïdes pendant 14 jours ou moins. 9,44 % du groupe expérimental ont reçu un diagnostic d’hypogonadisme après cinq ans, et 4,85 % ont bénéficié d’une thérapie à la testostérone pour le traitement de l’hypogonadisme. Ces résultats sont cliniquement significatifs et les cliniciens devraient être attentifs à la nécessité éventuelle d’un contrôle de la testostérone chez les patients masculins auxquels on a prescrit un traitement opioïde prolongé. Cependant, la prévalence de l’hypogonadisme dans cette population était beaucoup plus faible que ne le laissaient supposer des études antérieures à petite échelle qui suggéraient un taux d’incidence pouvant atteindre 90 %.
9. Remise en question, sur la base de données probantes, des hypothèses conventionnelles des médecins sur les opioïdes
9.1 Hypothèses conventionnelles sur l’efficacité relative
Outre la simple réduction de la dépendance des médecins à l’égard des opiacés pour l’analgésie à court terme, des études réexaminent actuellement les moyens et les méthodes d’administration de l’analgésie dans le cadre d’interventions médicales courantes. L’un des principaux domaines d’investigation est l’administration d’une analgésie lors d’une césarienne. En 2020, Sharpe et al. ont examiné l’efficacité de l’hydromorphone en remplacement de la morphine, qui a longtemps été considérée comme l’analgésie intrathécale de référence pour les césariennes. L’étude n’a montré aucune différence entre l’analgésie produite par l’hydromorphone et celle produite par la morphine. Bien que l’hydromorphone présente également un potentiel de dépendance important, le résultat a confondu les chercheurs qui pensaient que la morphine produirait une meilleure anesthésie. Bien qu’elle ne compare pas directement la morphine à un médicament non opioïde, cette étude est susceptible de susciter d’autres enquêtes mettant à l’épreuve des hypothèses de longue date sur les applications des opioïdes à la douleur chirurgicale. En outre, l’hydromorphone entraîne moins de somnolence subjective et permet une récupération postopératoire plus rapide.
9.2 Hypothèses conventionnelles sur les indications secondaires
Peu après l’introduction de l’opioïde mépéridine en Amérique latine, Calderyo-Barcia a décrit une association entre la mépéridine et la contractilité utérine. Par conséquent, la mépéridine a toujours été utilisée dans les hôpitaux d’Amérique latine pour indiquer la dystocie. Un essai contrôlé randomisé de 23 mois publié en 2004 par Sosa et al. a démontré qu’il n’y avait pas de différence entre la durée moyenne du travail des patientes souffrant de dysotcie qui avaient reçu de la mépéridine et celles qui n’en avaient pas reçu. Par conséquent, l’étude a conclu que la dystocie était une indication inappropriée de la mépéridine.
10. Remplacer les applications courantes de la thérapie opioïde aiguë par des alternatives non opioïdes
Le moyen le plus direct de réduire la prévalence de la dépendance aux opioïdes est de réexaminer objectivement l’efficacité des médicaments non opioïdes par rapport aux opioïdes dans les contextes où les médicaments opioïdes peuvent être considérés comme appropriés. Un essai contrôlé randomisé réalisé en 2017 a examiné les résultats de 416 patients se présentant aux urgences pour des douleurs aiguës modérées à sévères aux extrémités justifiant une radiographie. Chang et al. ont comparé l’efficacité de 400 mg d’ibuprofène et de 1000 mg d’acétaminophène par rapport à des combinaisons équivalentes d’oxycodone, d’hydrocodone ou de codéine, chacune avec une dose appropriée d’acétaminophène. L’étude a conclu qu’il n’y avait pas de différence significative entre les combinaisons étudiées, ce qui suggère que la combinaison de 400 mg d’ibuprofène et de 1000 mg d’acétaminophène est un substitut raisonnable pour les douleurs aiguës courantes aux urgences (69).
11. Examen des applications localisées des opioïdes
La recherche d’une plus grande analgésie avec des doses plus faibles d’opioïdes s’étend à l’expérimentation d’applications plus localisées d’anesthésiques opiacés.
11.1 Administration épidurale ou intraveineuse de la mépéridine
Dès 1994, Paech et al. ont démontré que la voie d’administration des médicaments opiacés pouvait réduire l’exposition globale aux analgésiques opiacés. Cette étude présentait une variante de l’anesthésie contrôlée par le patient dans laquelle l’opioïde mépéridine était administré par voie péridurale au lieu d’être administré par voie intraveineuse. Les patients recevant une administration épidurale ont eu besoin de doses de mépéridine significativement plus faibles que ceux recevant une administration intraveineuse.
D’autres études de longue date ont démontré qu’après une administration épidurale, la disponibilité plus concentrée du métabolite actif de la mépéridine, la nor-mépéridine, crée un plus grand risque de toxicité hors cible pour le CNS, comme les crises d’épilepsie, les tremblements, l’hyperréflexie myoclonique et l’agitation. Cependant, lorsqu’ils sont identifiés, ces effets secondaires sont réversibles en 1 à 2 jours avec un traitement de soutien et l’administration de benzodiazépines (tableau 1).
[TABLEAU 1]
12. Conclusions
Dans le contexte de la crise actuelle des opioïdes, le potentiel de dépendance des opiacés est en train d’être réconcilié après de nombreuses décennies de surconsommation. Heureusement, avec l’avènement de la médecine factuelle, la disponibilité croissante des données grâce aux dossiers électroniques et aux systèmes de gestion de la facturation, ainsi que la disponibilité croissante d’outils précis de biologie moléculaire, les médecins sont bien placés pour réévaluer les indications conventionnelles d’opioïdes spécifiques. En outre, ces outils permettent aux médecins de tester de nouvelles applications opioïdes et non opioïdes, ainsi que de nouvelles voies d’administration pour obtenir une analgésie supérieure tout en réduisant le fardeau des effets secondaires systémiques et les risques de dépendance et d’accoutumance. Lorsque le cancer en phase terminale et d’autres douleurs chroniques justifient une thérapie opioïde continue pour des douleurs intenses, de nouveaux protocoles pour l’utilisation en rotation des thérapies opioïdes et de leurs adjuvants non opioïdes peuvent maintenir une efficacité analgésique supérieure sur de plus longues périodes de temps.
La pratique procédurale actuelle permet aux médecins d’appliquer les opioïdes sur des zones de distribution de plus en plus étroites et ciblées. À l’avenir, il est prévisible que les progrès des techniques cellulaires et moléculaires permettront de surmonter les éternels problèmes de dépendance, d’accoutumance et de tolérance, car les médecins auront la possibilité de manipuler la prévalence de récepteurs cellulaires spécifiques. De même, la manipulation de ces récepteurs est prometteuse pour limiter les divers effets systémiques hors cible chez les patients nécessitant un traitement opioïde chronique. Les médecins doivent être les gardiens de l’utilisation des opioïdes et ne les utiliser qu’en cas de nécessité.
